November 2021 Case Study
by Dillon Nussbaum, MD
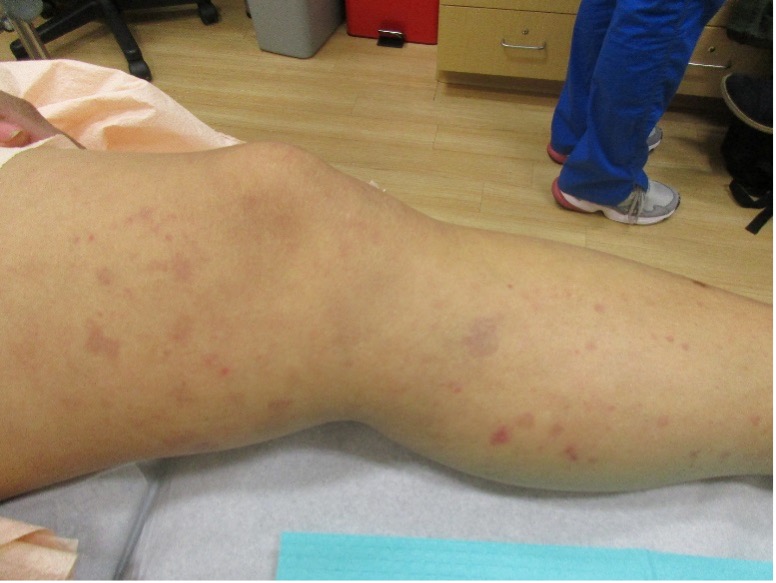
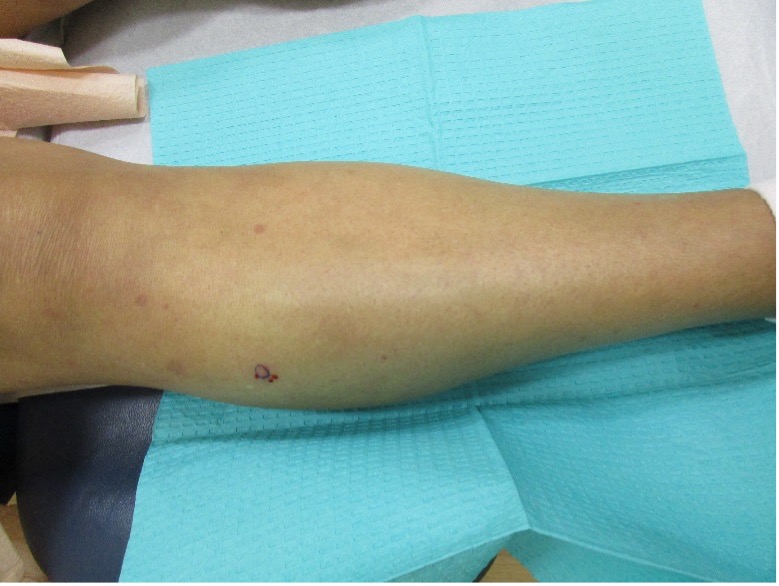
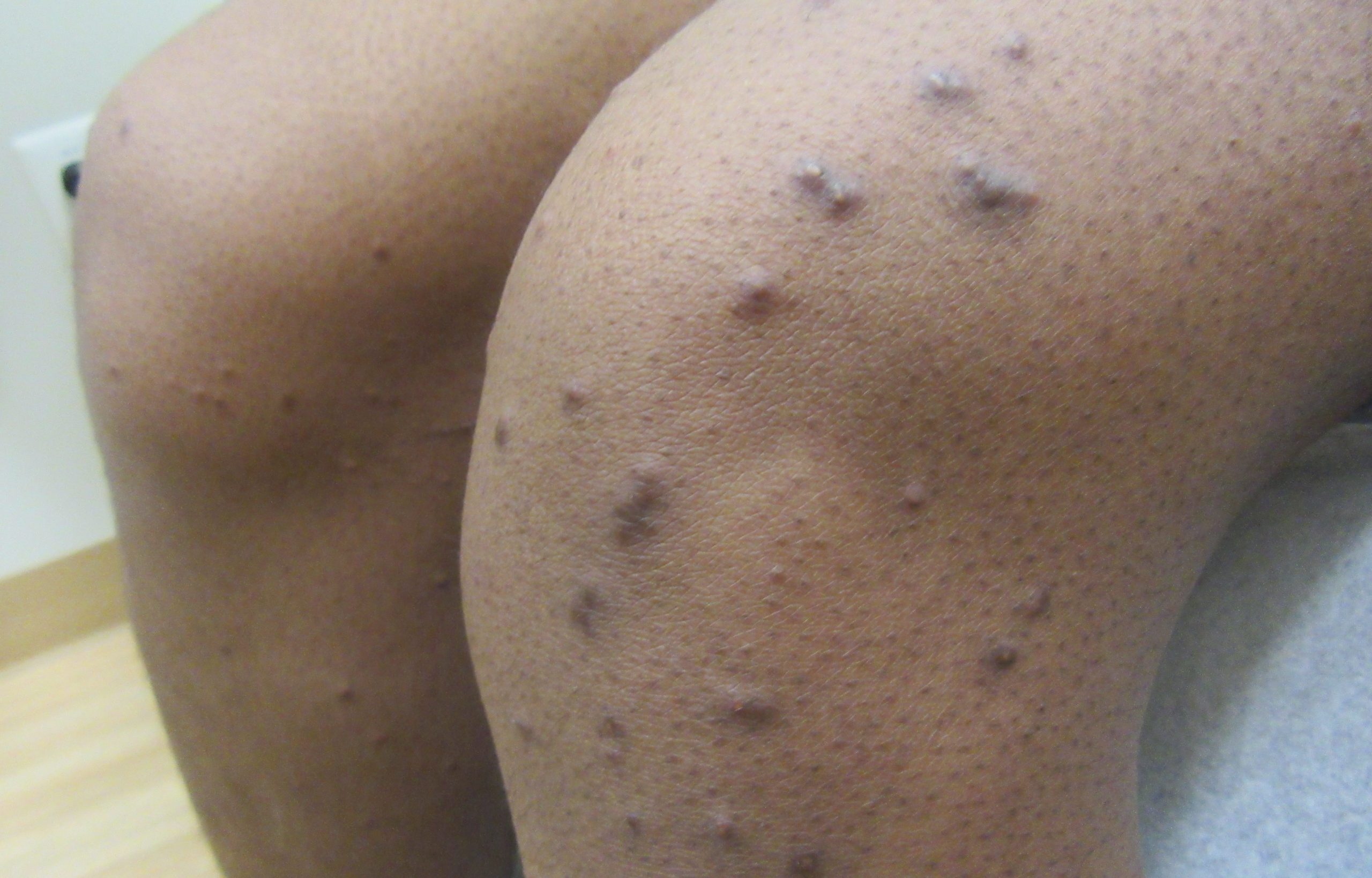
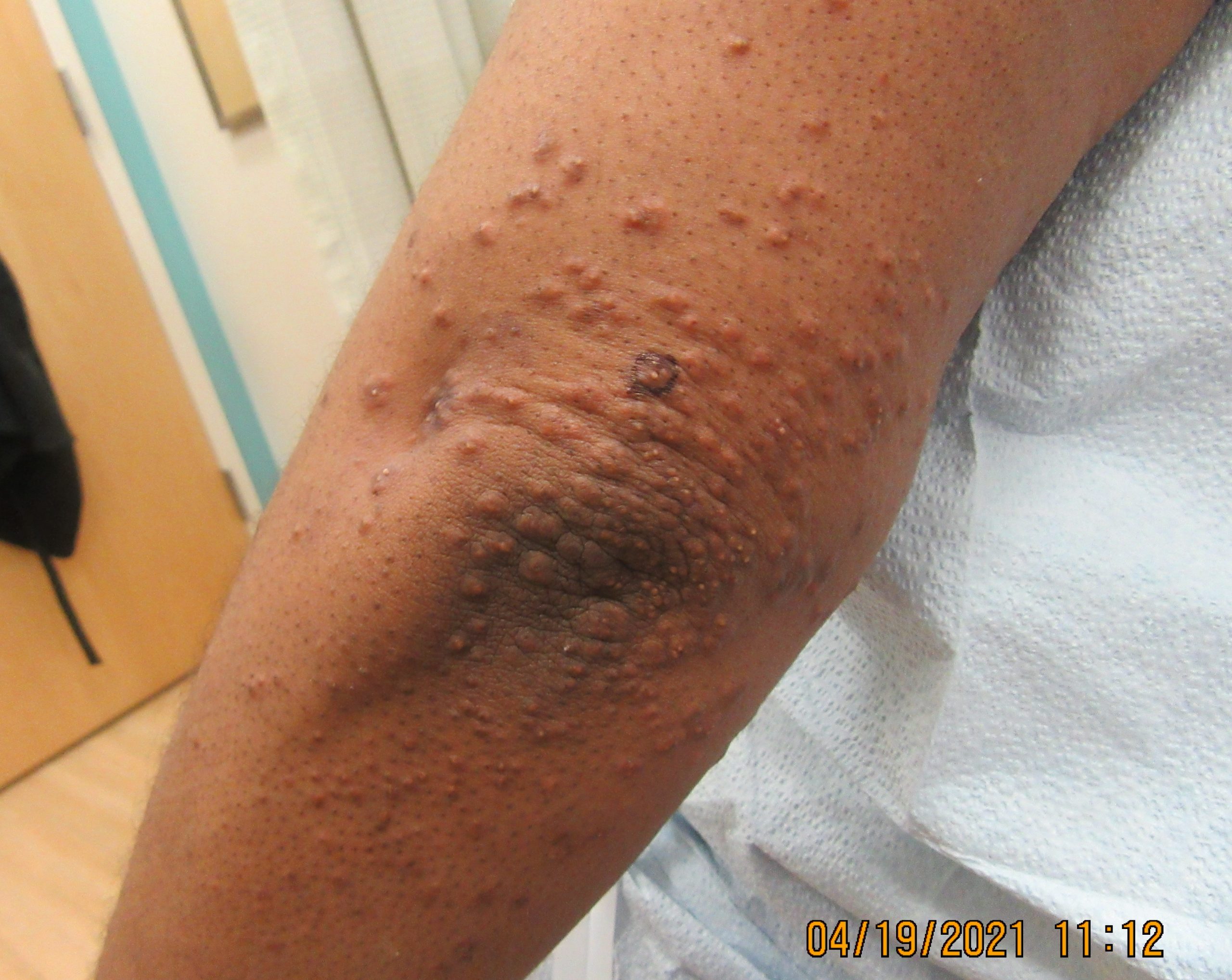
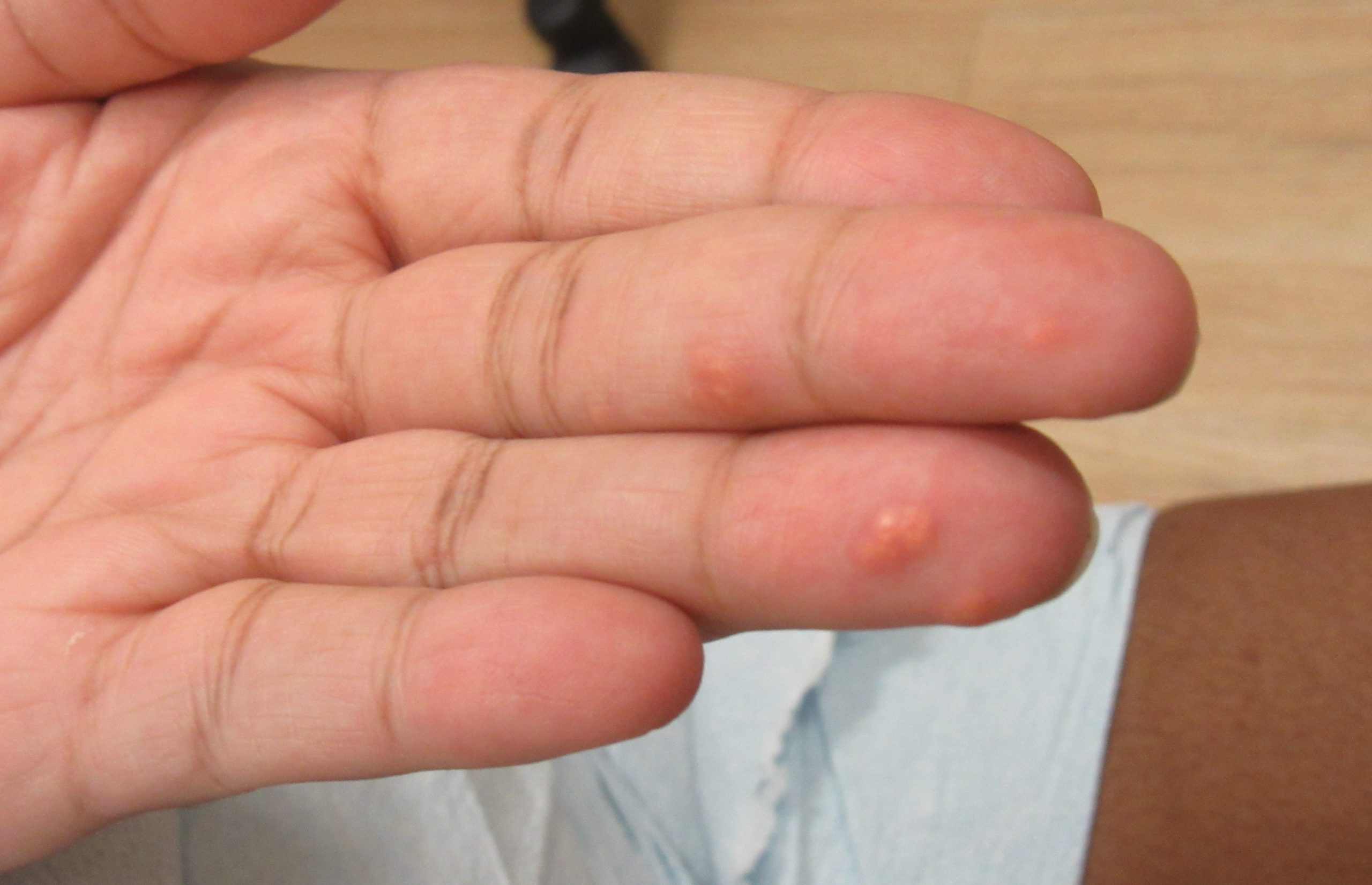
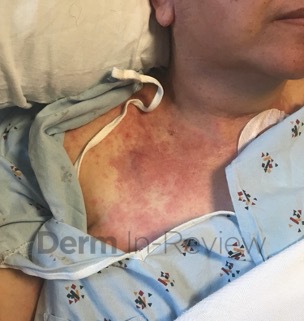
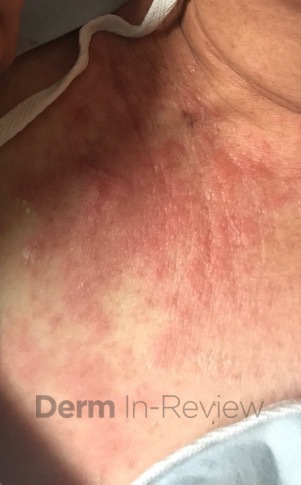
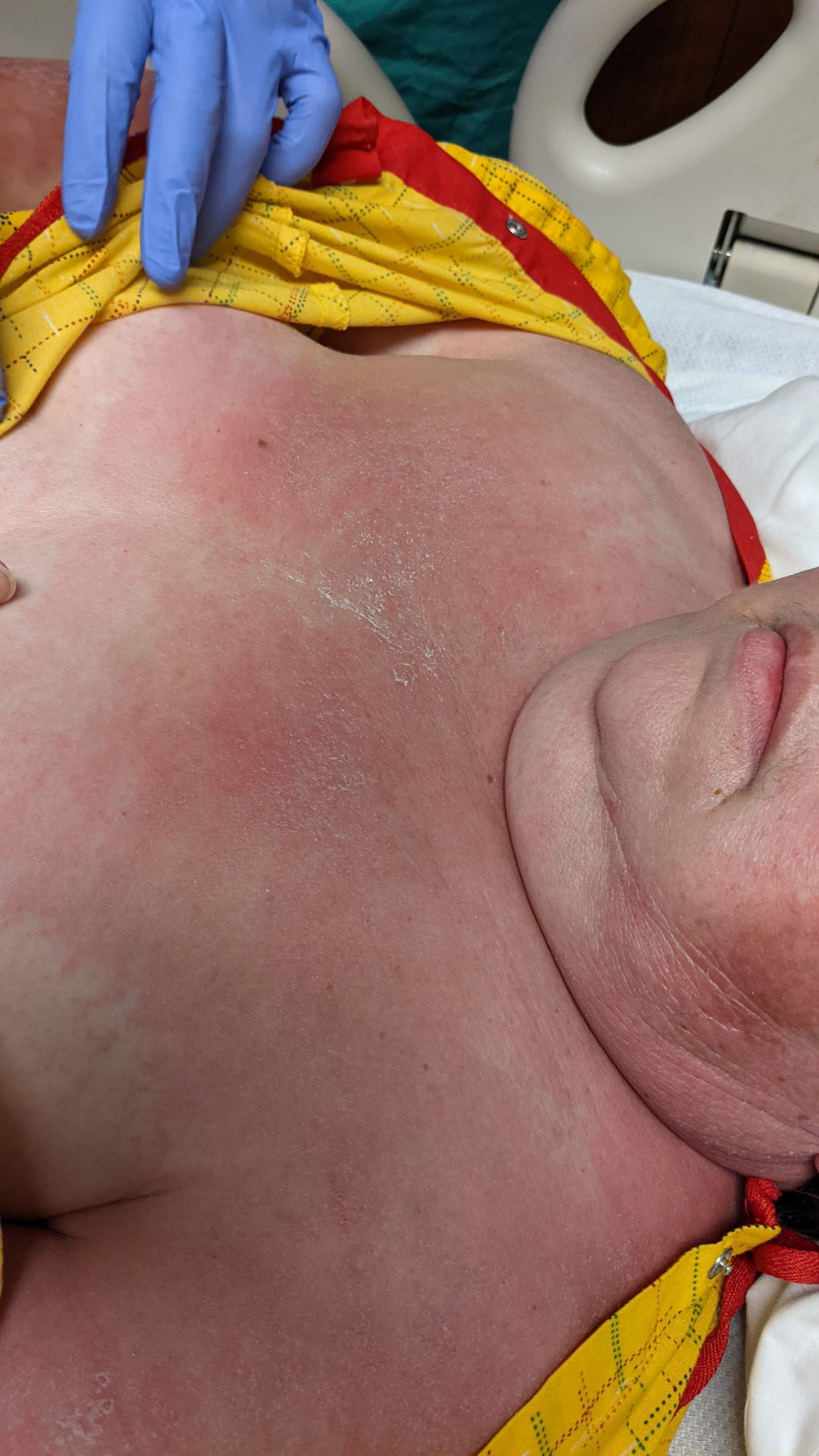
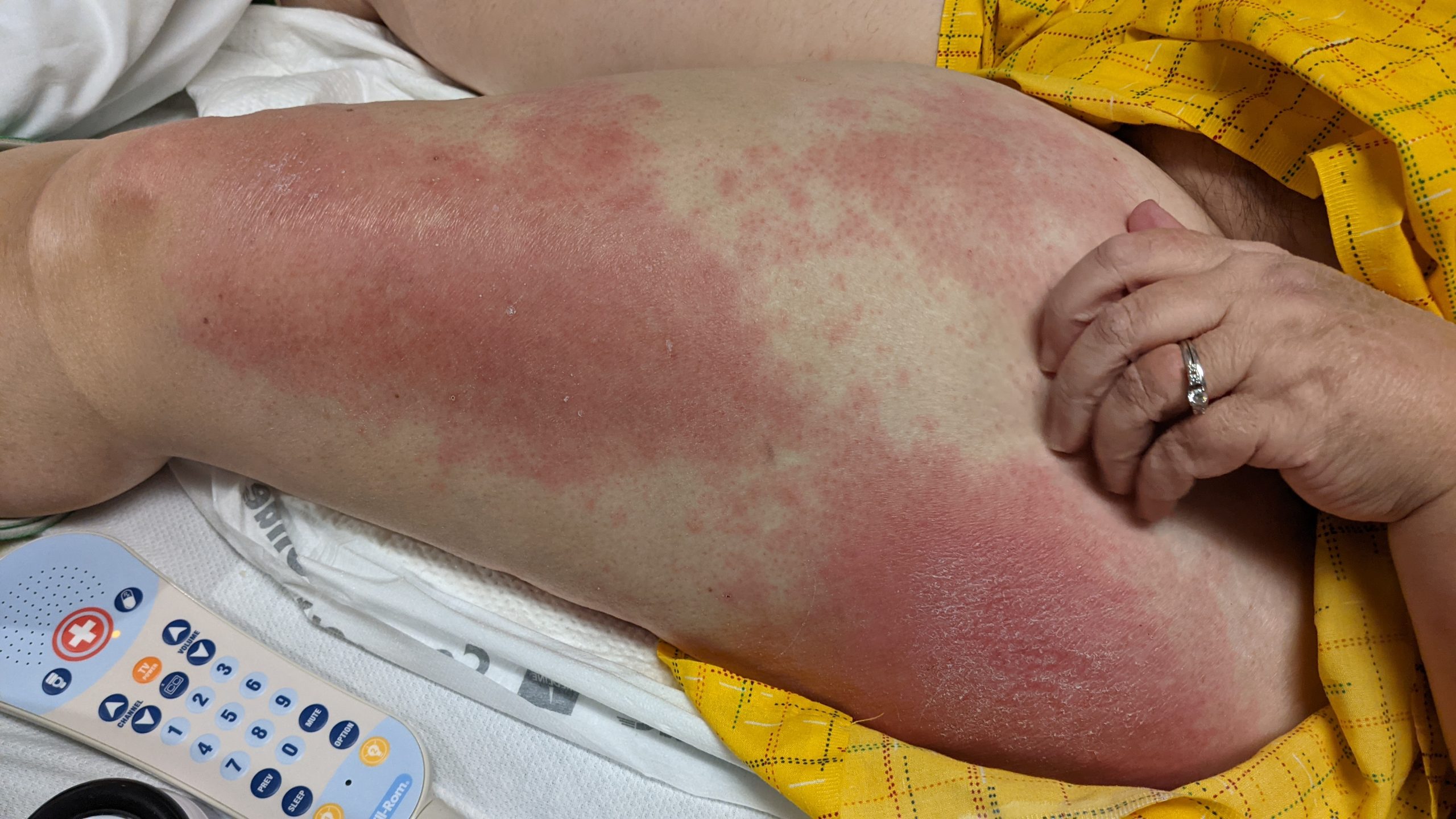
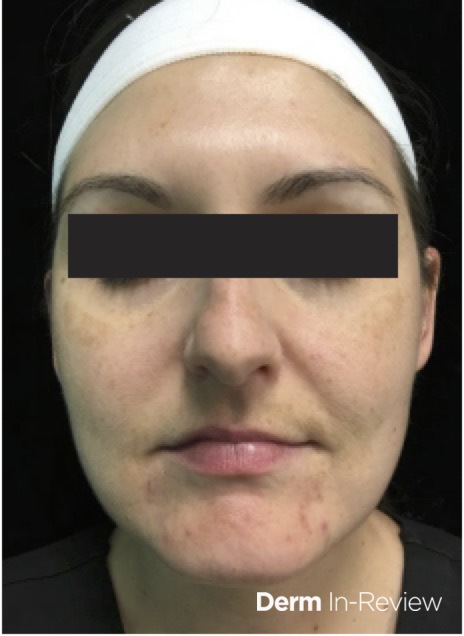
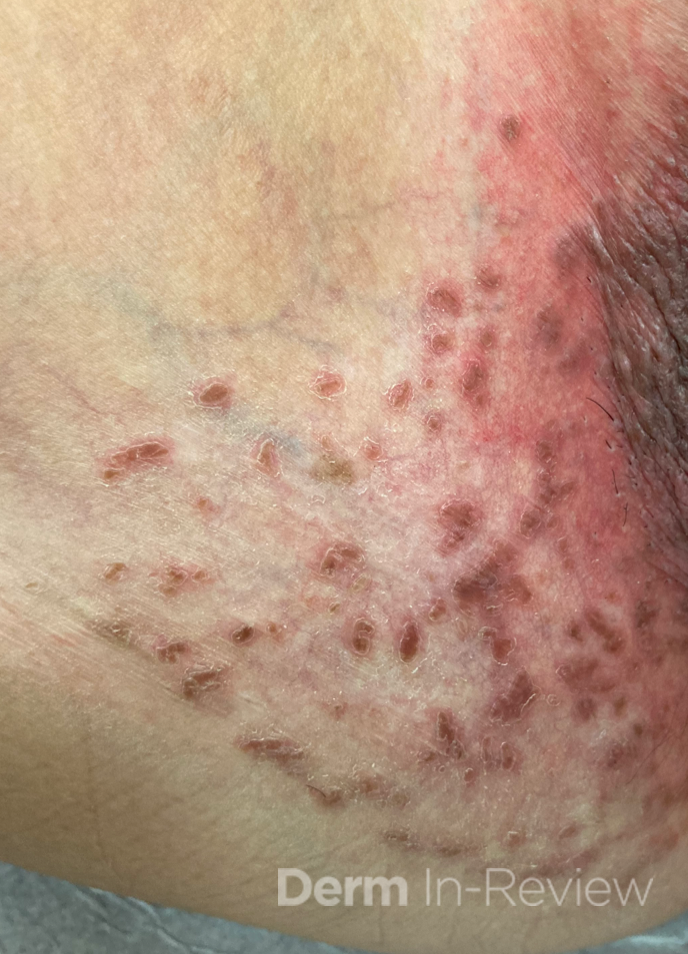
A 31-year-old female with no past medical history presents for evaluation of the pictured lesions of the right upper thigh. The lesions have been present since birth and occasionally pruritic. She was previously treated unsuccessfully with electrodessication, topical sulfur, and combination betamethasone-gentamycin in another country. She denies any systemic symptoms and has no personal or family history of skin conditions other than the pictured lesions.
Immunohistochemical staining of these lesions would most likely be positive for all of the following EXCEPT:
A.) LYVE-1
B.) S-100
C.) Prox1
D.) Podoplanin
E.) VEGFR-3
Correct answer: B
Explanation:
The history and image provided are most consistent with a microcystic lymphatic malformation, previously known as lymphangioma circumscriptum. Microcystic lymphatic malformations are usually a result of congenital abnormalities in the development of superficial lymphatic channels, but they can be acquired following trauma, radiation, or a malignancy. They typically appear as grouped clear, yellow, or red appearing papules and/or vesicles resembling frog spawn.
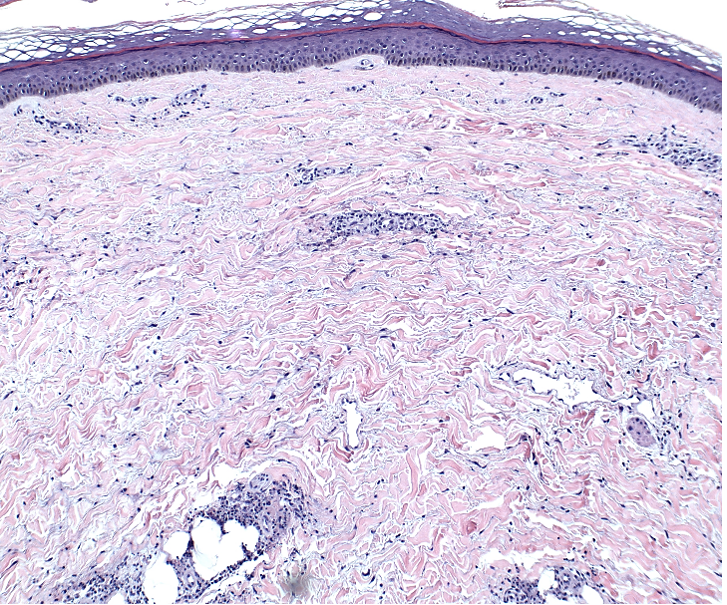
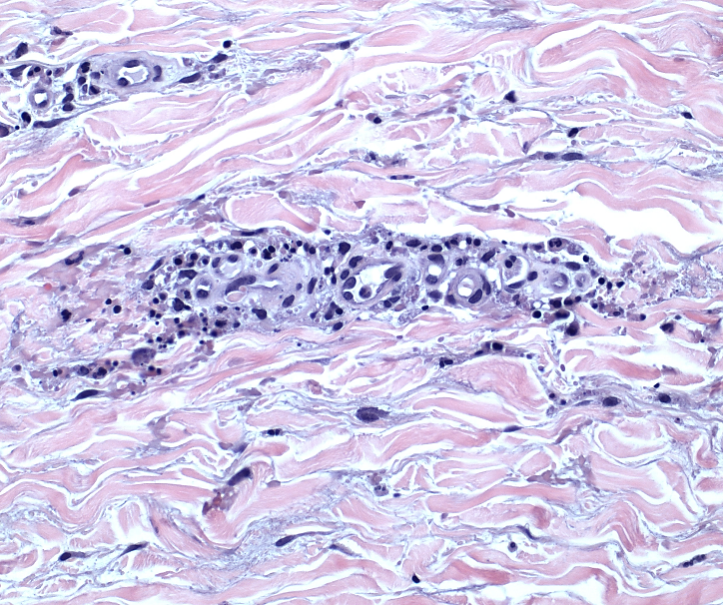
Histopathology of microcystic lymphatic malformations reveals enlarged and abnormal appearing lymphatic vessels with smooth muscle cells lining thin walled endothelium. The lymphatic endothelium can be identified by immunohistochemical staining for LYVE1, Prox1, Podoplanin, and VEGFR-3.1
LYVE-1, Prox1, podoplanin (D2-40), and VEGFR-3 are all immunohistochemical stains for lymphatic tissue and therefore will be positive when staining the pictured lesions. These markers are also useful for identifying angiosarcoma, kaposi sarcoma, lymphangiosarcoma, and mesothelioma among other vascular and lymphatic based conditions or tumors. However, the remaining answer, S-100, is generally positive in in melanoma, nerve sheath tumors, schwannomas, clear cell sarcoma of soft tissue, various histiocytic tumors, glial tumors and myoepithelial tumors.2,3
There are no standard treatment guidelines for microcystic lymphatic malformations. Depending on location and presentation, treatment can consist of a combination of surgical excision, sclerotherapy, laser therapy, hyfrecation, cryotherapy, and topical sirolimus. Macrocystic lymphatic malformations are seen commonly in turner syndrome as cystic hygromas on the neck; microcystic lymphatic malformations are less commonly associated with syndromic pictures.4
References
- Davis M, van der Hilst J. Mimickers of urticaria: Urticarial vasculitis and autoinflammatory diseases. J Allergy Clin Immunol Pract. 2018. 6(4):1162-1170.
- Bolognia, Jean L., MD; Schaffer, Julie V., MD; Cerroni, Lorenzo, MD. Dermatology, Fourth Edition. 2018.
- Gu SL, Jorizzo JL. Urticarial vasculitis. Int J Womens Dermatol. 2021 Jan 29;7(3):290-297. doi: 10.1016/j.ijwd.2021.01.021. PMID: 34222586; PMCID: PMC8243153.
- Magro C, Nuovo G, Mulvey J, Laurence J, Harp J, Crowson AN. The skin as a critical window in unveiling the pathophysiologic principles of COVID-19. Clin Dermatol. 2021 Jul 22. doi: 10.1016/j.clindermatol.2021.07.001. Epub ahead of print. PMCID: PMC8298003.
- Jara LJ, Navarro C, Medina G, Vera-Lastra O, Saavedra MA. Hypocomplementemic urticarial vasculitis syndrome. Curr Rheumatol Rep. 2009 Dec;11(6):410-5. doi: 10.1007/s11926-009-0060-y. PMID: 19922730.