
August 2024 Case Study
Author: Nagasai Adusumilli, MD, MBA1
- Department of Dermatology, The George Washington University School of Medicine and Health Sciences
Patient History
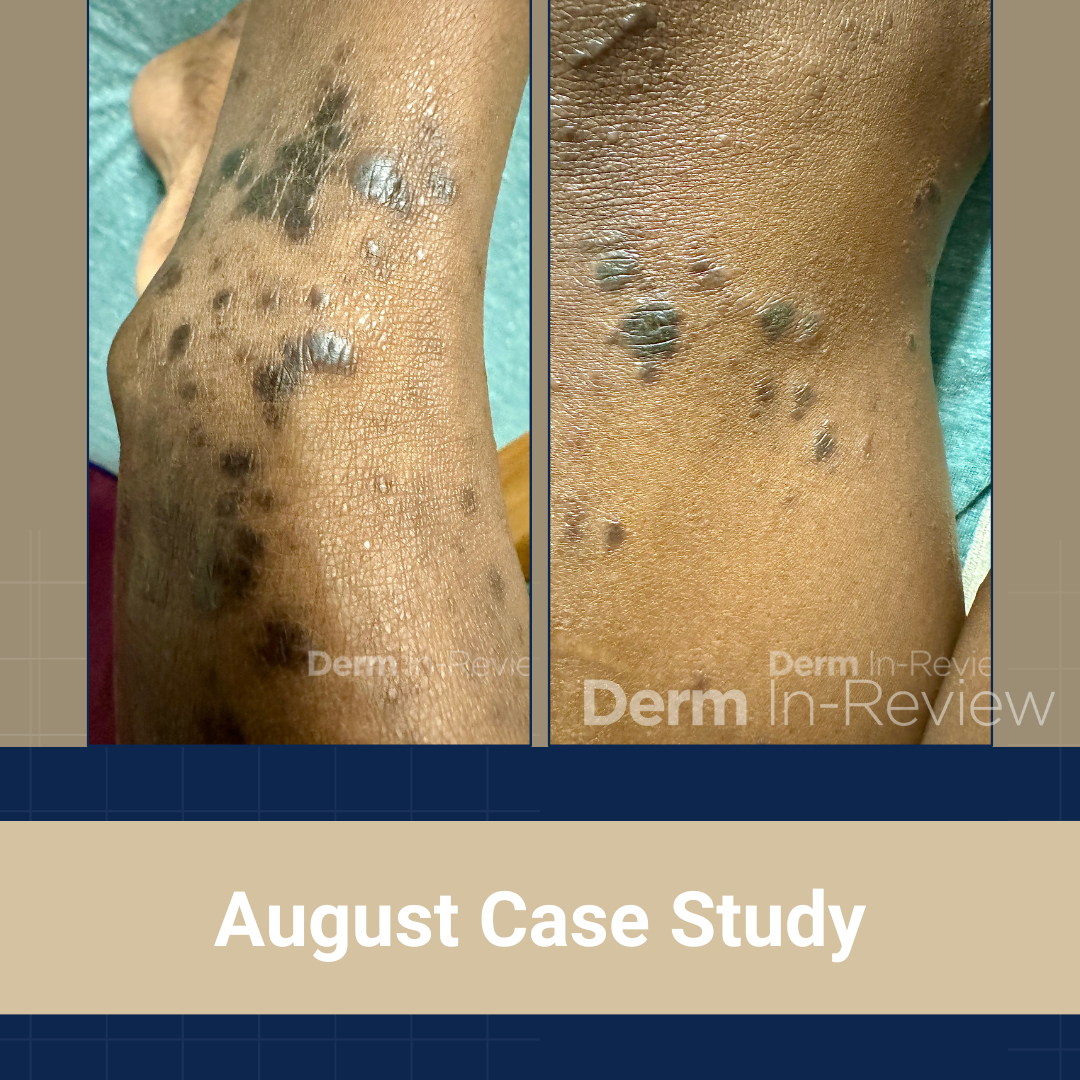
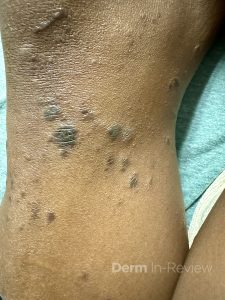
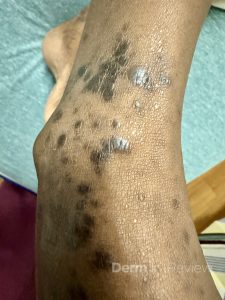
A 64-year-old female with a medical history of chronic microcytic anemia presented to the clinic with 1 month of intensely pruritic discolored lesions on the lower legs. They have been increasing in number and extending onto the lower thighs, prompting her visit. She denies any changes to her chronic medical history or any daily medications. Skin findings of the bilateral extensor knees and ankles are shown in Figures 1 and 2. No primary lesions were appreciated in the oral cavity or genital region. Preliminary bloodwork was pertinent for a negative HIV and syphilis screen.
Based on the clinical presentation, which phenomenon/sign is illustrated, and in what other condition can this be appreciated?
A.) Meyerson; pemphigus vulgaris
B.) Asboe-Hansen; hemangioma
C.) Darier’s; dermatomyositis
D.) Gorlin’s; keratosis pilaris atrophicans faciei
E.) Koebner; hemangioma
F.) Samitz; pemphigus vulgaris
G.) Hertoghe’s; mastocytoma
H.) Koebner; vitiligo
I.) Darier’s; neurofibromatosis type I
J.) Gorlin’s; Ehlers-Danlos
K.) Meyerson; dermatomyositis
L.) Samitz; pityriasis rubra pilaris
M.) Hertoghe’s; lichen nitidus
Correct Answer: H
Explanation/Literature review:
This patient has lichen planus, with the typical morphology of well-demarcated, flat-topped, dark brown to violaceous papules in clusters.1 Classic distributions include distal lower extremities, as with this patient, wrists, forearms, and lower back.1 The linear configuration seen in both figures, in the context of the intense symptom of pruritus, is suggestive of the Koebner phenomenon or isomorphic response.1,2 A cluster of lesions arranged in a linear pattern result from injury to previously uninvolved skin, including from scratching, arthropod bites, piercing, tattoos, and surgery.2 True Koebnerization can be seen in a wide range of conditions, including but not limited to psoriasis, lichen nitidus (M), cutaneous small vessel vasculitis, juvenile rheumatoid arthritis, vitiligo, Darier’s disease (keratosis follicularis), and pityriasis rubra pilaris (L).2,3 Answer choice H correctly identifies both the illustrated phenomenon and another condition in which it can be observed.
Incorrect answer choices:4
Although eponyms in dermatology are slowly phasing out, we remain in a transition period in which distinct findings and conditions are still commonly referred to by historical names in both clinical practice and academic writing. As they build their dermatology lexicon, trainees must still be familiar with these eponymous signs.
Meyerson phenomenon (A, K) – halo of eczematous dermatitis of scale and erythema overlying melanocytic lesions5
Asboe-Hansen sign (B) – enlargement or extension of a vesicle or bullae to surrounding skin by applying downward finger pressure. Can be seen in various vesicobullous conditions, including pemphigus vulgaris (A, F).
Darier’s sign (C, I) – Rubbing induces urticaria, swelling, or vesiculation due to histamine release. Classically seen in a mastocytoma (G). Can also be seen in leukemia cutis, juvenile xanthogranuloma, and Langerhans cell histiocytosis.
Gorlin’s sign (D, J) – Patients with Ehlers-Danlos can extend their tongue to touch the nasal tip. While answer choice J is has appropriate associations, this does not fit the vignette.
Hemangioma (B, E) – The Kasabach-Merritt phenomenon can be seen in tufted angiomas and kaposiform hemangioendotheliomas. Platelets are trapped in vascular lesions, leading to rapid growth of the lesion and thrombocytopenia.
Samitz sign (F, L) – Frayed or ragged nail folds characteristic of nail changes in dermatomyositis (C, K).
Hertoghe’s sign (G, M) – Loss of lateral eyebrow hair that can be seen a wide range of conditions, including keratosis pilaris atrophicans faciei (D), alopecia areata, follicular mucinosis, discoid lupus, and trichotillomania.
Neurofibromatosis type I (I) – Crowe’s sign refers to the axillary freckling that can be part of the diagnostic criteria for NF1.
References:
- Shiohara T & Mizukawa Y. Chapter 11 – Lichen Planus and Lichenoid Dermatoses. In: Dermatology. 5th Edition. Elsevier; 189-209.
- High WA, Tomasini CF, Argenziano G, et al. Chapter 0 – Basic Principles of Dermatology. In: Dermatology. 5th Edition. Elsevier; 1-99.
- Sharma RK, Gupta M, Gulati A. Koebner phenomenon in classic juvenile onset pityriasis rubra pilaris. Indian Dermatol Online J. 2019 Jul-Aug;10(4):469-470. PMID: 31334074.
- Madke B, Nayak C. Eponymous signs in dermatology. Indian Dermatol Online J. 2012 Sep;3(3):159-65. PMID: 23189246.
- Nicholls DS, Mason GH. Halo dermatitis around a melanocytic naevus: Meyerson’s naevus. Br J Dermatol. 1988 Jan;118(1):125-9. PMID: 3342172.