June 2023 Case Study
by Sapana Desai, MD & Kamaria Nelson, MD
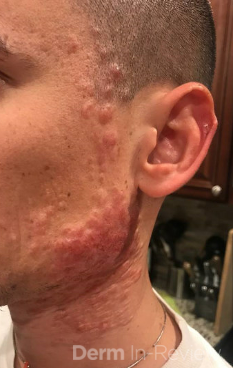
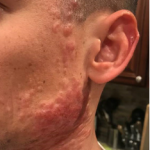
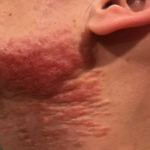
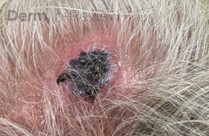
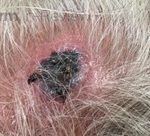
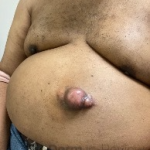
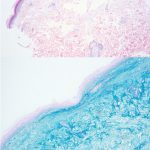
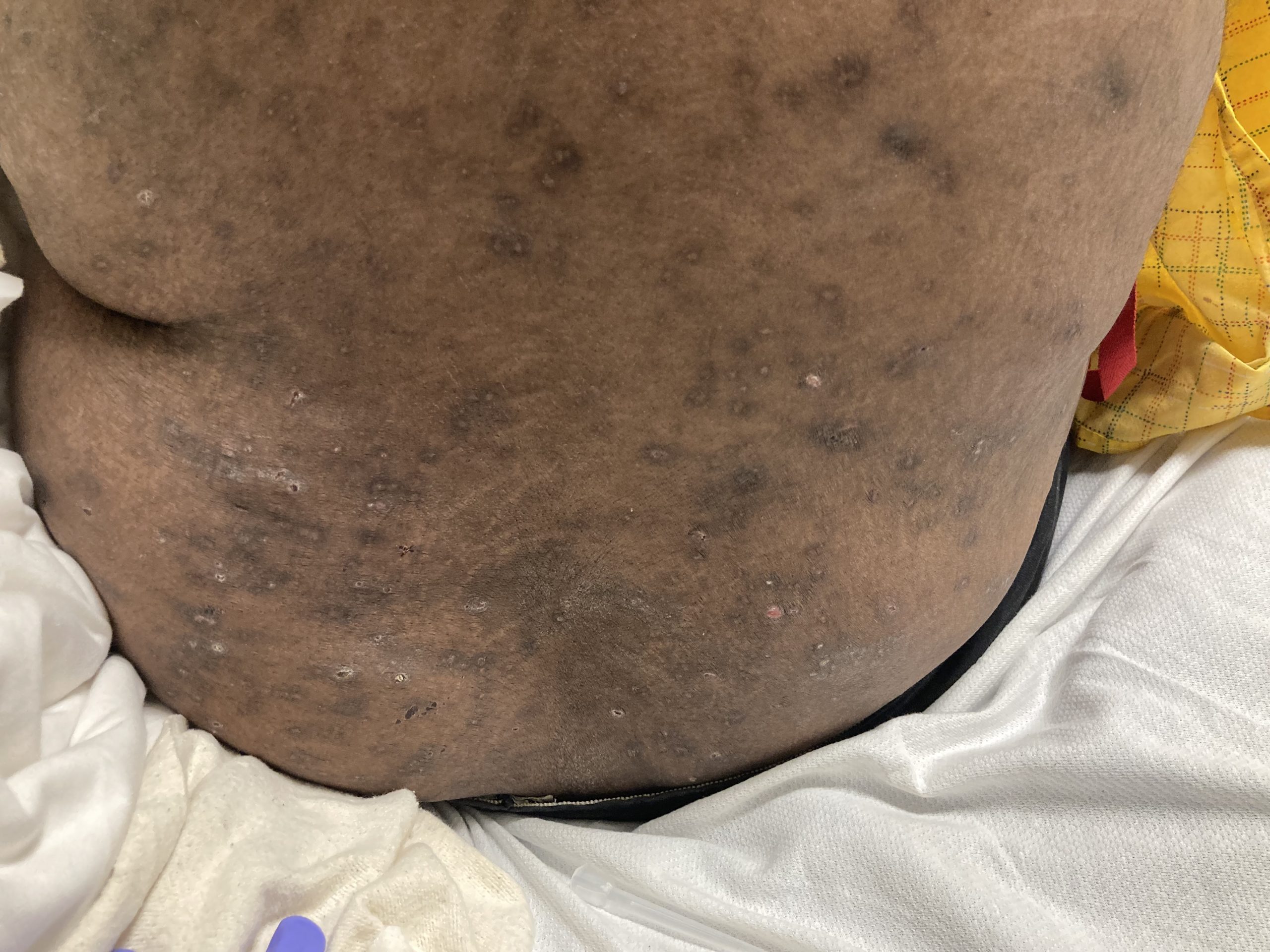
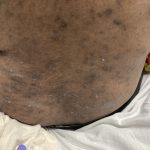
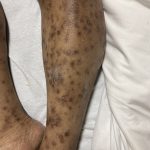
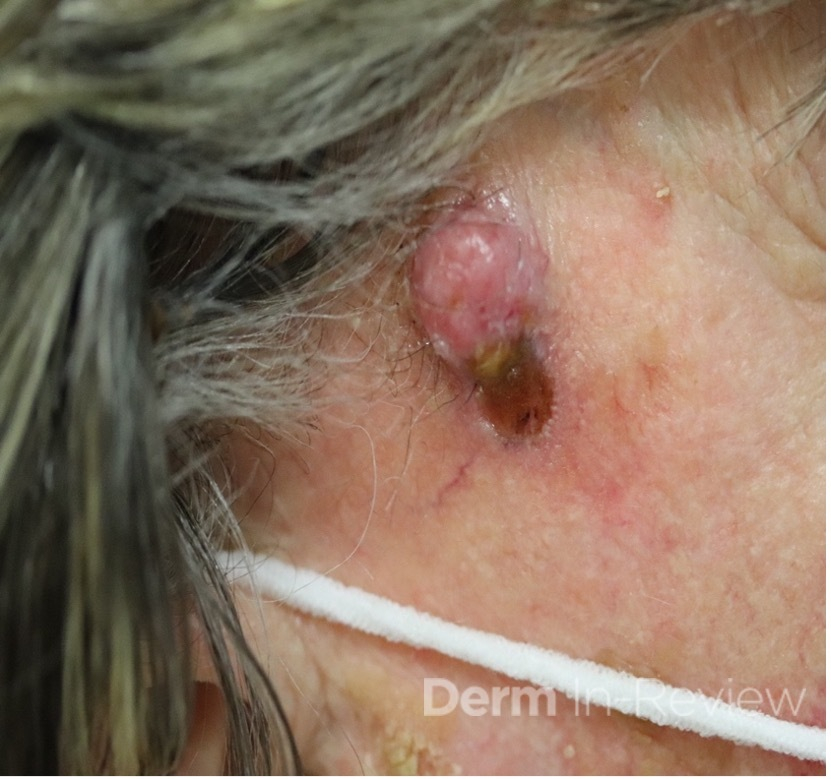
A 93-year-old Caucasian female patient with significant past medical history of atrial fibrillation, chronic obstructive pulmonary disease, and rheumatoid arthritis presents with a single, painless, cherry red colored, glistening dome-shaped nodule on her right mid temporal scalp that appeared two months ago. Although asymptomatic, the palpable nodule rapidly evolved to measuring 3.5 x 4.5 x 4cm. Physical examination findings are shown in Figure 1.
Which of the following immunohistochemical marker is strongly associated with the patient’s diagnosis?
A) HHV8 LNA-1
B) CK20
C) Factor XIIIa
D) PHLDA1
E) CD31
Correct Answer: B.) CK20
Explanation of Correct Answer:
This elderly patient’s clinical presentation, which entails spontaneous emergence of a rapidly growing, asymptomatic reddish to violaceous spherical tumor with a smooth, shiny surface and a soft to turgid elastic consistency localized to the face- a region with greatly increased ultraviolet (UV) exposure, is highly suspicious for Merkel Cell Carcinoma (MCC).
MCC is a rare and aggressive cutaneous malignancy of neuroendocrine origin that most frequently presents with head and neck distribution and is characterized by high rates of local recurrence and nodal metastasis; five-year survival rates of all stages are 50-to-60%.1 MCC arises in individuals of advanced age with peak incidence of 75 years of age, and predominantly affects Caucasian males when compared to women and other ethnic groups. While etiology of MCC remains elusive, clonal integration of the Merkel cell DNA double-stranded polyomavirus (MCPyV) into the host genome, or UV-mediated genetic aberrations affecting the tumor suppressor p53 and RB1 pathways as well as epigenetic factors that activate oncogenes, are accepted theories in carcinogenesis.1,2,3 Other culprit risk factors encompass chronic UV exposure, lymphoproliferative diseases, and iatrogenic or pathological immunosuppression.3
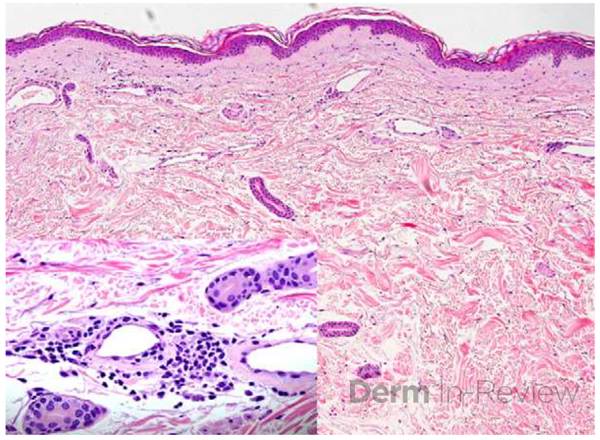
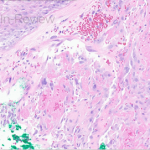
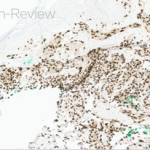
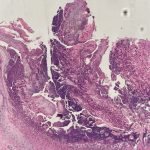
Histopathologic assessment of MCC will reveal infiltration of the dermis and subcutaneous tissue- often sharing a connection with overlying epidermis and adnexal tissue, and exhibit sheets and nests of crowded basaloid cells with a finely granular “salt and pepper” chromatic pattern, indistinct nucleoli, and scant cytoplasm.1 Mitoses and extensive necroses are common and reflect the detrimental nature of these tumor. Because histomorphology of MCC is rarely specific, immunohistochemistry is imperative for definitive diagnosis and can be confirmed via detection of CK20 (B), typically identified in perinuclear granules; metastasis of Small Cell Lung Carcinoma (SCLC) can be excluded if thyroid transcription factor 1 (TTF-1) is negative. In unusual circumstances if staining patterns for MCC are atypical or CK20 fails to be expressed, additional immunohistochemical markers including Pancytokeratin (AE1/3), chromogranin A, neuron specific enolase (NSE), synaptophysin, INSM1, or N-CAM (CD56) may be used.1,4
Surgical modalities are most often utilized for primary MCC neoplasms, whether it be via wide local excisions (WLE) with 1-to-2cm margins, Mohs micrographic surgery (MMS), or excision with complete circumferential peripheral and deep margin assessment. In non-excisable circumstances and one-third of MCC cases with distant nodal and intra-abdominal metastases, immunotherapy with checkpoint inhibitors targeting programmed death receptor-1 (PD-1)/ programmed dead ligand-1(PD-L1) pathways have proved to be beneficial, preventing inhibition of T-cells and leading to increased immune system activity and antitumor responses. Current marketed immunotherapies for advanced stages of MCC include: Avelumab– a humanized anti-PD-L1 antibody (dose: 800mg IV q2weeks), Pembrolizumab– a humanized IgG4 antibody directed against PD-1 (dose: 2mg/kg q3weeks x 3 years), and Nivolumab- a fully human IgG4 antibody directed against PD-1 (dose: 240mg IV q2weeks).15
Explanation of Incorrect Answers:
Human Herpes Virus-8 Latency Associated Nuclear Antigen 1 (HHV8 LNA-1) (A) immunohistochemistry positivity is both sensitive and highly specific for diagnosis of Kaposi Sarcoma (KS), an angioproliferative mesenchymal malignant neoplasm caused by infection with Kaposi Sarcoma-Related Herpesvirus.5,6 Expression of HHV8 LNA-1 causes binding of p53 and suppression of apoptosis. Subsequent activation of NF-kB elicits up-regulation of VEGF and bFGF and consequent neoangiogenesis, prompting characteristic KS histomorphological features of dermal masses of spindle cells with extravasated red blood cells and cellular atypia.5 KS is classified into four epidemiological subtypes: 1) classic form- presenting increased predilection amidst elderly men of Mediterranean and Eastern European descent with lower extremity involvement, 2) endemic African form– occurring amongst pediatric populations with generalized lymph node involvement, 3) AIDS-related form- afflicting HIV positive male homosexuals with diffused dermal and systemic involvement, and 4) iatrogenic form- affecting immunosuppressed individuals with analogous dissemination and effects as the latter.5,6 While cutaneous findings in KS range from scattered pink to violaceous patches and papules to rapidly progressive, multicentric, ulcerated plaques and nodules with dissemination to visceral organs [eg. lungs and gastrointestinal system], lesions are often painful with associated lymphedema and secondary infection.6
Factor XIIIa (C) immunohistochemistry is a strongly positive staining marker for Dermatofibromas.7 Regarded as benign fibrohistocytic neoplasms, Dermatofibromas clinically present as asymptomatic, solitary, firm to hard, often hyperpigmented, slow-growing, plaque or cutaneous nodules with or without overlying dermal changes.7,8 The “dimple sign” is a characteristic finding where lateral inward digital pressure of the skin produces central dimpling over the lesion. Dermatofibromas occur in people of all ages with increased predilection for the extremities, and most commonly ensue spontaneously with no inciting event but in a fifth of all reported cases are attributed to a primary reactive process from history of local trauma, such as vaccination or an insect bite.8 Classical histopathological features consist of ill-defined intersecting bundles of spindle cells with collagen trapping.7,8 Several dermoscopic patterns of Dermatofibromas are described, of which the most common is central white scar-like patch with delicate pigmentary network at the periphery; other typical patterns include total homogenous pigmentation and irregular crypts associated with pseudofollicular openings.7,8 It is especially vital to differentiate Dermatofibromas from its similar-appearing, more advanced and aggressive counterpart, Dermatofibrosarcoma protuberans [DFSP], with the latter staining positive for CD-34 and negative for FactorXIIIa.8
PHLDA1 (D) along with stromal CD34 and CD10 immunohistochemistry positivity is often used to delineate Trichoblastomas from Basal Cell Carcinoma as the two diagnoses commonly share histopathological features, including the presence of fissures between the epithelium and spindled-fibroblast follicular stroma, epithelial nests, and cells in a palisading arrangement on the tumor periphery.9,11 Trichoblastomas are rare, benign dermal tumors arising from aberrant proliferation of primitive follicular germinate cells. They clinically present as asymptomatic, solitary, skin-colored, slow-growing nodules that measure less than 2 cm in diameter on the face or scalp. Overlying epidermis is alopecic, thin, and hyperpigmented, and occasionally exhibit superficial telangiectasias and ulcerations.10 Trichoblastomas may emerge sporadically, in association with hereditary disease including Brooke-Spiegler disease and Brooke-Fordyce syndrome, or within a nevus sebaceous, and typically affect adults of both genders within their fifth and sixth decades of life. More often than not, Trichoblastomas have favorable prognosis, with low incidence of progression, recurrence, or association with malignancy.9,10,11
Immunohistochemistry positivity for CD31 (E) as well as for other vascular markers including CD34 and factor VIII antigen are suggestive for diagnosis of pyogenic granuloma, a benign vascular tumor that arises within cutaneous and mucosal tissues affecting patients of all ages.12 Although staining is often unnecessary given the characteristic clinical history and histological architecture, atypical lesions may necessitate its use. Histopathology shows highly vascularized proliferation of granulation tissue, lobular arrangements of capillary vessels and proliferating endothelial cells delineated by fibrous septae, scattered fibroblasts, and a variegated inflammatory infiltrate.12,13 Pyogenic granuloma appears as a solitary, red, pedunculated papule that is greatly friable, and exhibits rapid exophytic growth. Lesions often undergo ulceration and can bleed profusely with even minor trauma.14 While the pathophysiology is not fully elucidated, imbalance of pro-angiogenic and anti-angiogenic factors, hormonal influences with increased estrogen and progesterone levels during pregnancy, reactive granulation tissue from negligible injuries, and iatrogenic use of specific medications are all plausible contributors.12,13,14
References
- Becker, JC., Beer, A., DeTemple, V., Eigentler, T., Flaig, M., Gambichler, T., Grabbe, S., Holler, U., Klumpp, B., Lang, S., Pfohler, C., Posch, C., Prasad, V., Schlattmann, P., Ter-Nedden, J., Terheyden, P., Thoms, K., Vordermark, D., Ugurel, S. S2K Guideline- Merkel Cell Carcinoma [MCC, Neuroendrocrine Carcinoma of the Skin]- Update 2022. Journal of the German Society of Dermatology. 2023; 21(3): 305-320.
- Hernandez, L., Mohsin, N., Yaghi, M., French, F., Dreyfuss, I., Nouri, K. Merkel Cell Carcinoma: An Updated Review of Pathogenesis, Diagnosis, and Treatment Options. Dermatologic Therapy. March 2022; 35(3).
- MD, Y., THakuria, M. MD. Merkel Cell Carcinoma Review. Hematology/Oncology Clinics of North America. February 2019; 33(1): 39-52.
- Brady, M., Spiker, A. Merkel Cell Carcinoma of the Skin. 2022.
- Biship, B., Lynch, D. Kaposi Sarcoma. 2022
- Etemad, S., Dewan, A. Kaposi Sarcoma Updates. Dermatologic Clinics. October 2019; 37(4): 505-517.
- Singh, S., Patra, S., Bhari, N. Dermatofibroma Over the Face. Indian Dermatology Online Journal. Jan-Feb 2019; 10(1): 94-95.
- Myers, D., Fillman, E. Dermatofibroma. 2022.
- Schukow, C., Ahmed, A. Trichoblastoma and Trichoepithelioma. StatPearls. 2022.
- Patel, P., Nawrocki, S., Hinther, K., Khachemoune, A. Trichoblastomas Mimicking Basal Cell Carcinoma: The Importance of Identification and Differentiation. Cureus. May 2020; 12(5).
- Cazzato, G., Cimmino, A., Colagrande, A., Arezzo, F., Lospalluti, L., Sablone, S., Lettini, T., Resta, L., Ingravallo, G. The Multiple Faces of Nodular Trichoblastoma: Review of the Literature with Case Presentation. Dermatopathology. 2021; 8(3): 265-270.
- Wollina, U., Langner, D., Franca, K., Gianfaldoni, S., Lotti, T., Tchernev, G. Pyogenic Granuloma- A Common Benign Vascular Tumor with Variable Clinical Presentation: New Findings and Treatment Options. Open Access Macedonian Journal of Medical Sciences. July 2017; 5(4): 423-426.
- Komakech, D., Ssenkumba, B. Pyogenic Granuloma. New England Journal of Medicine. November 2022; 387(21): 1979.
- Sarwal, P., Lapumnuaypol, K. Pyogenic Granuloma. 2022.
- Gauci, M., Aristei, C., Becker, J. Garbe, J., Lebbe, C. Diagnosis and Treatment of Merkel Cell Carcinoma: European Consensus-Based Interdisciplinary Guideline- Update 2022. European Journal of Cancer. August 2022; 17(1): 203-231.