December 2021 Case Study
by Azam Qureshi, MD
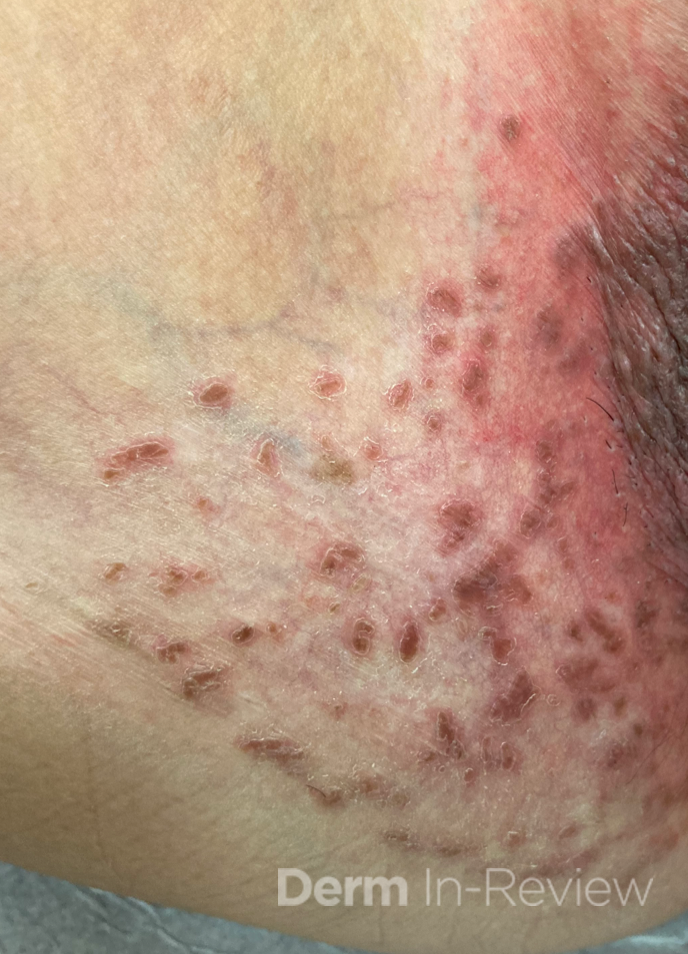
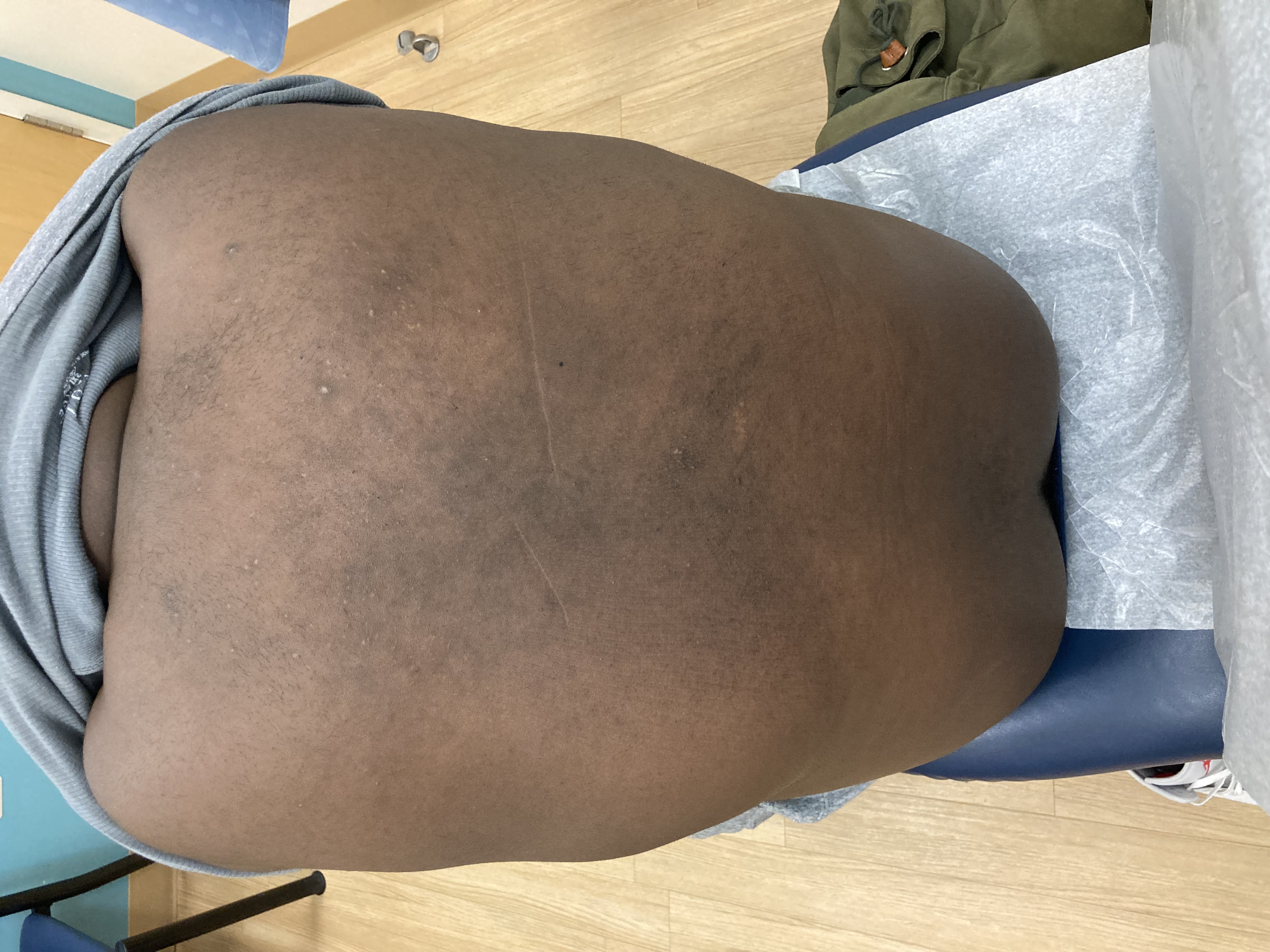
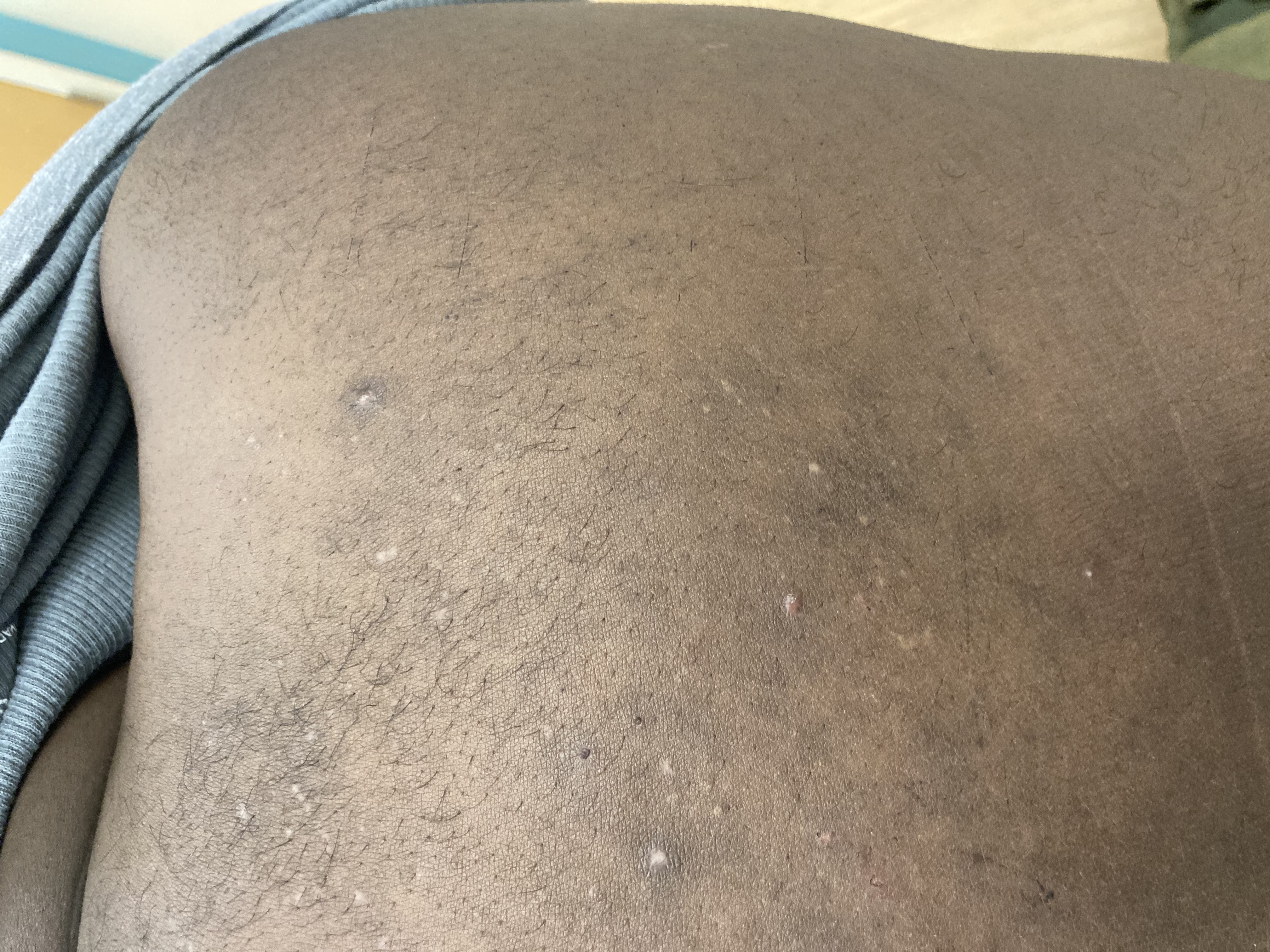
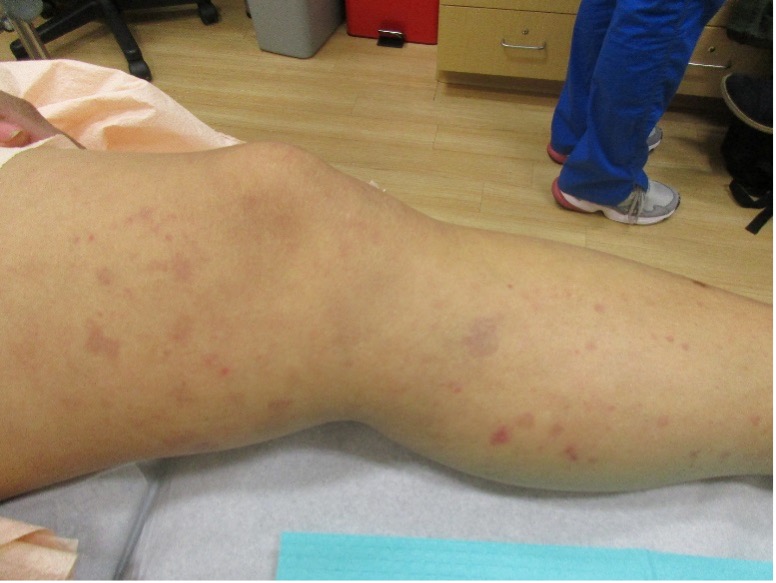
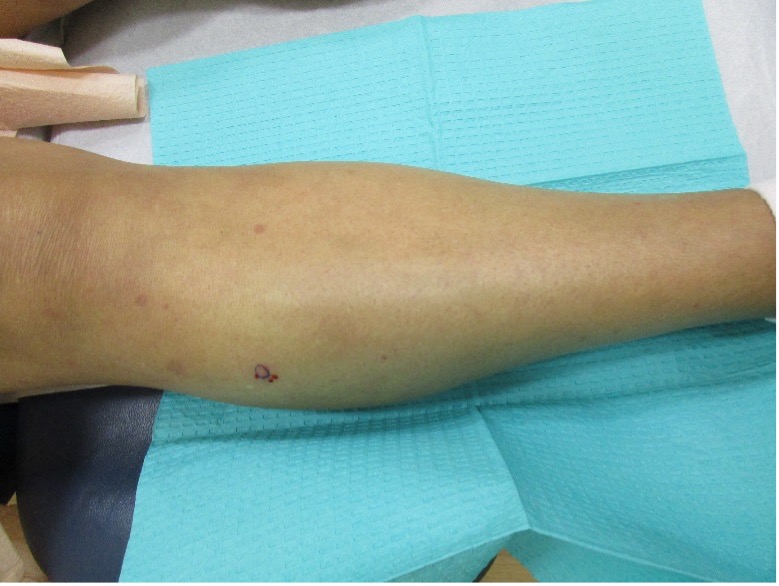
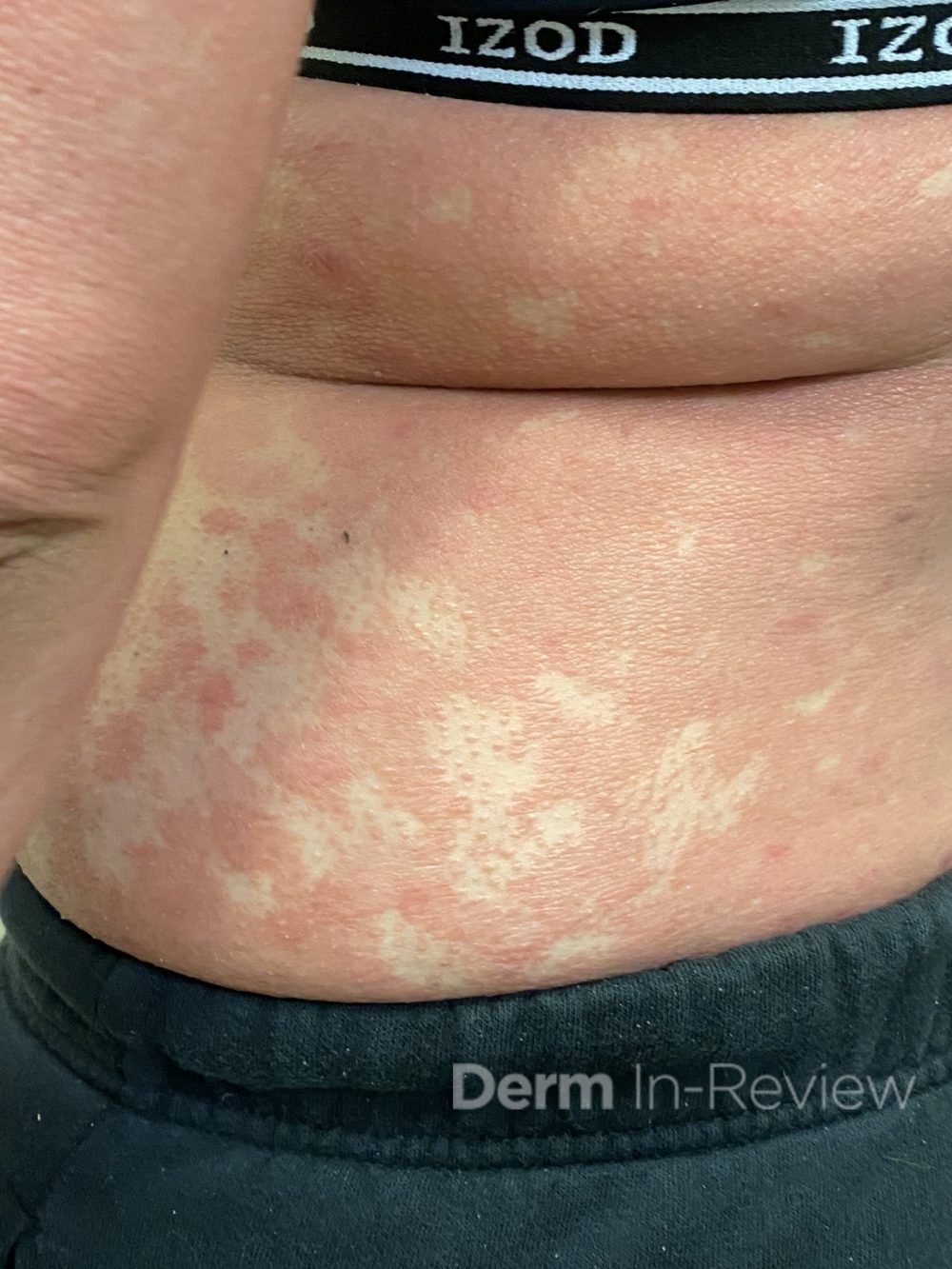
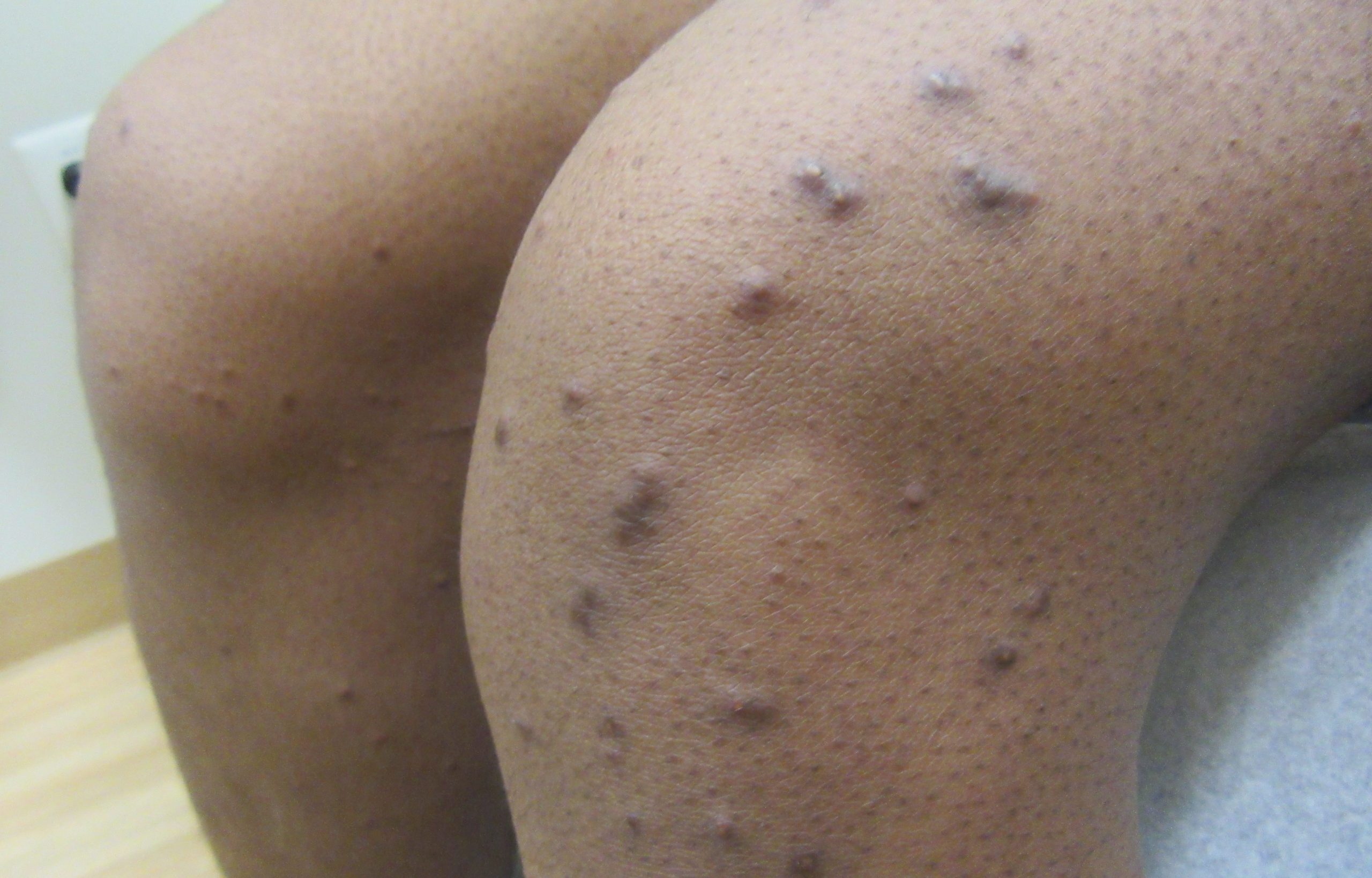
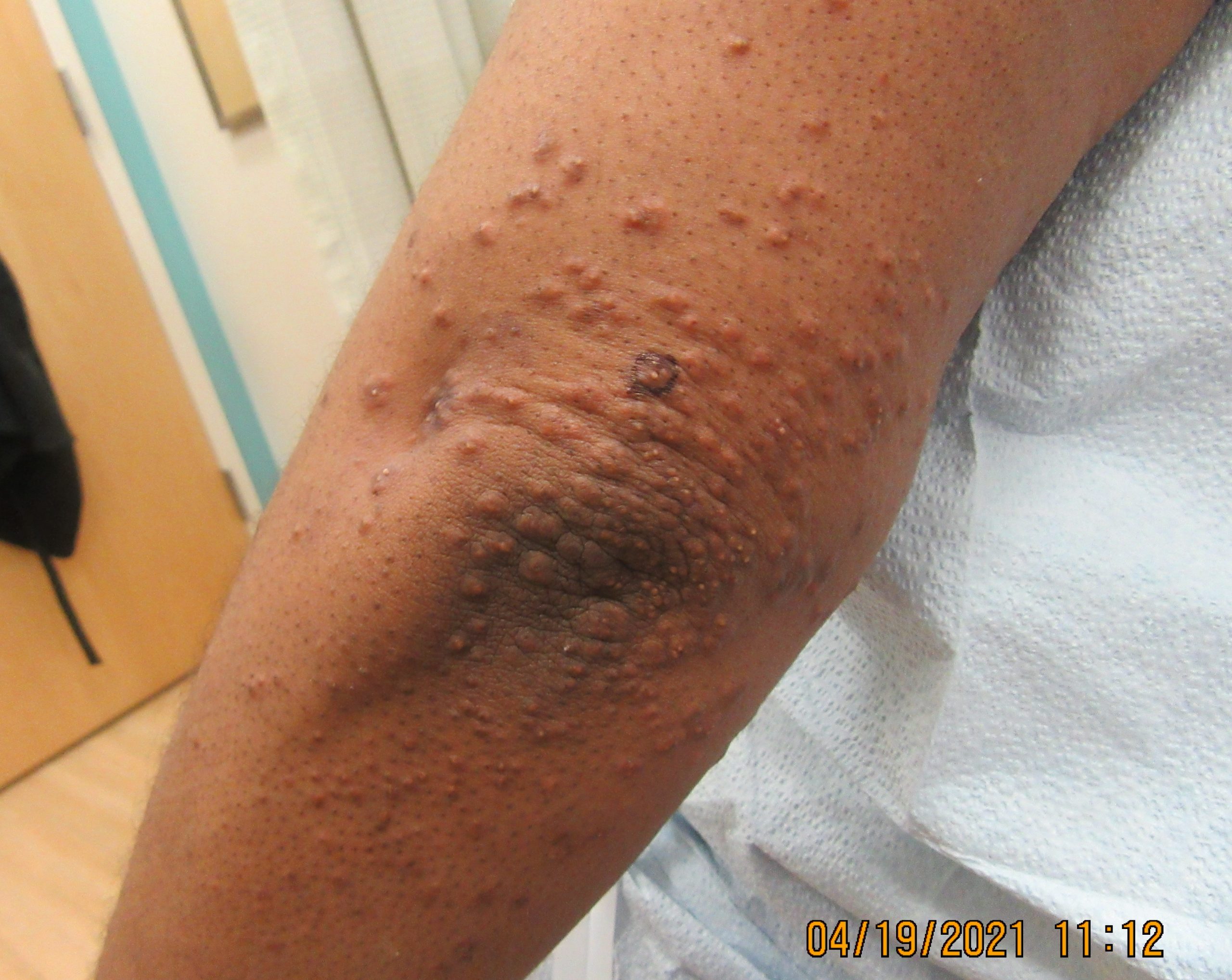
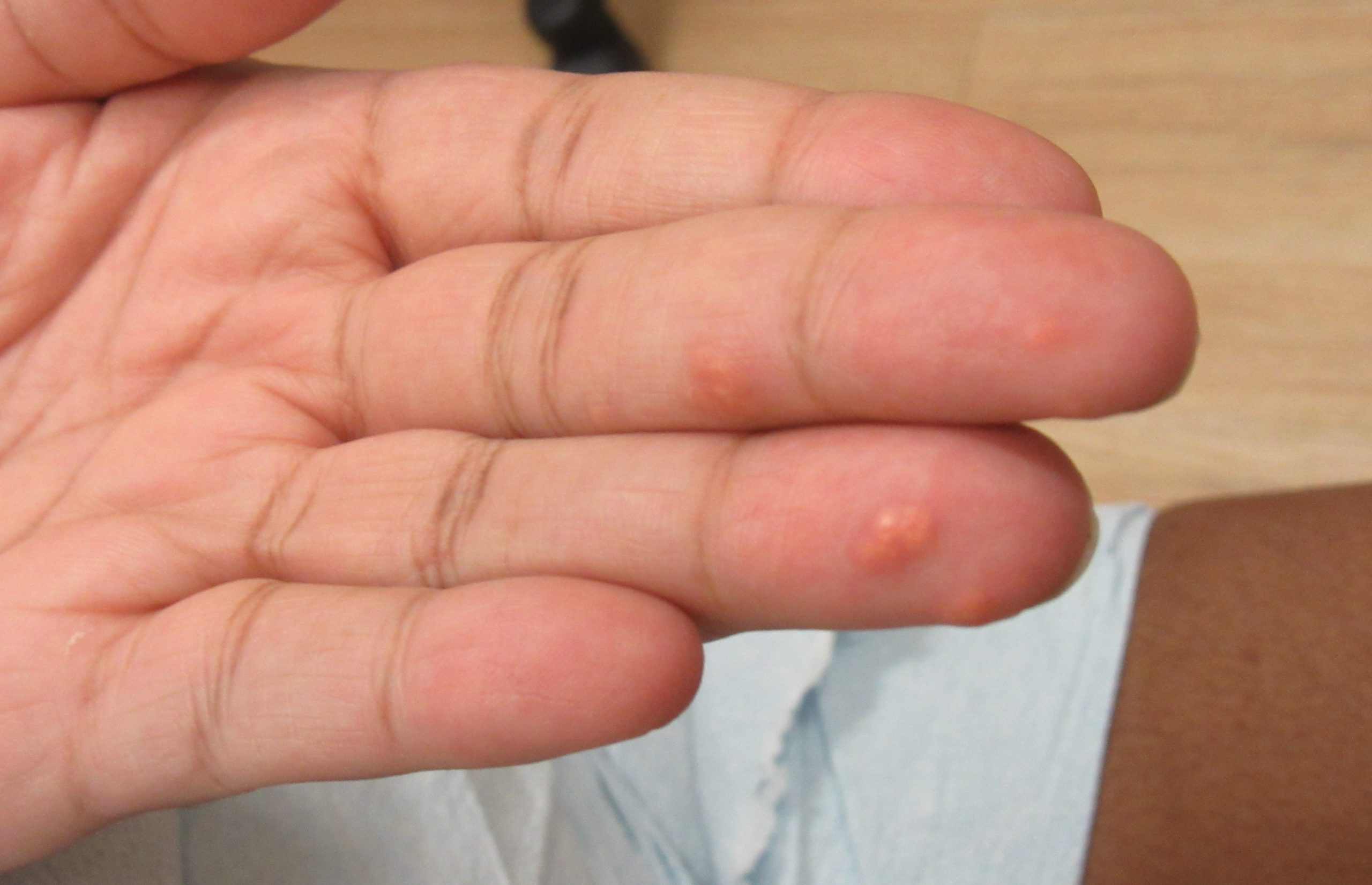
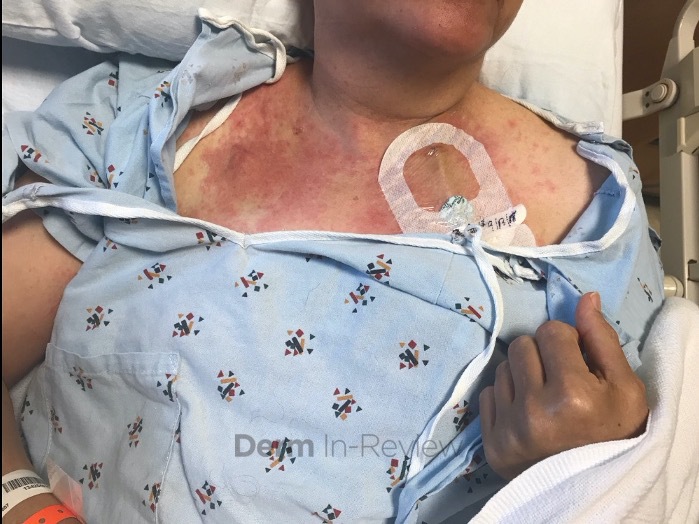
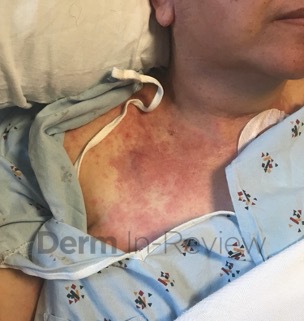
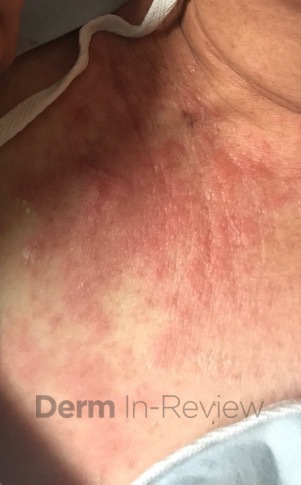
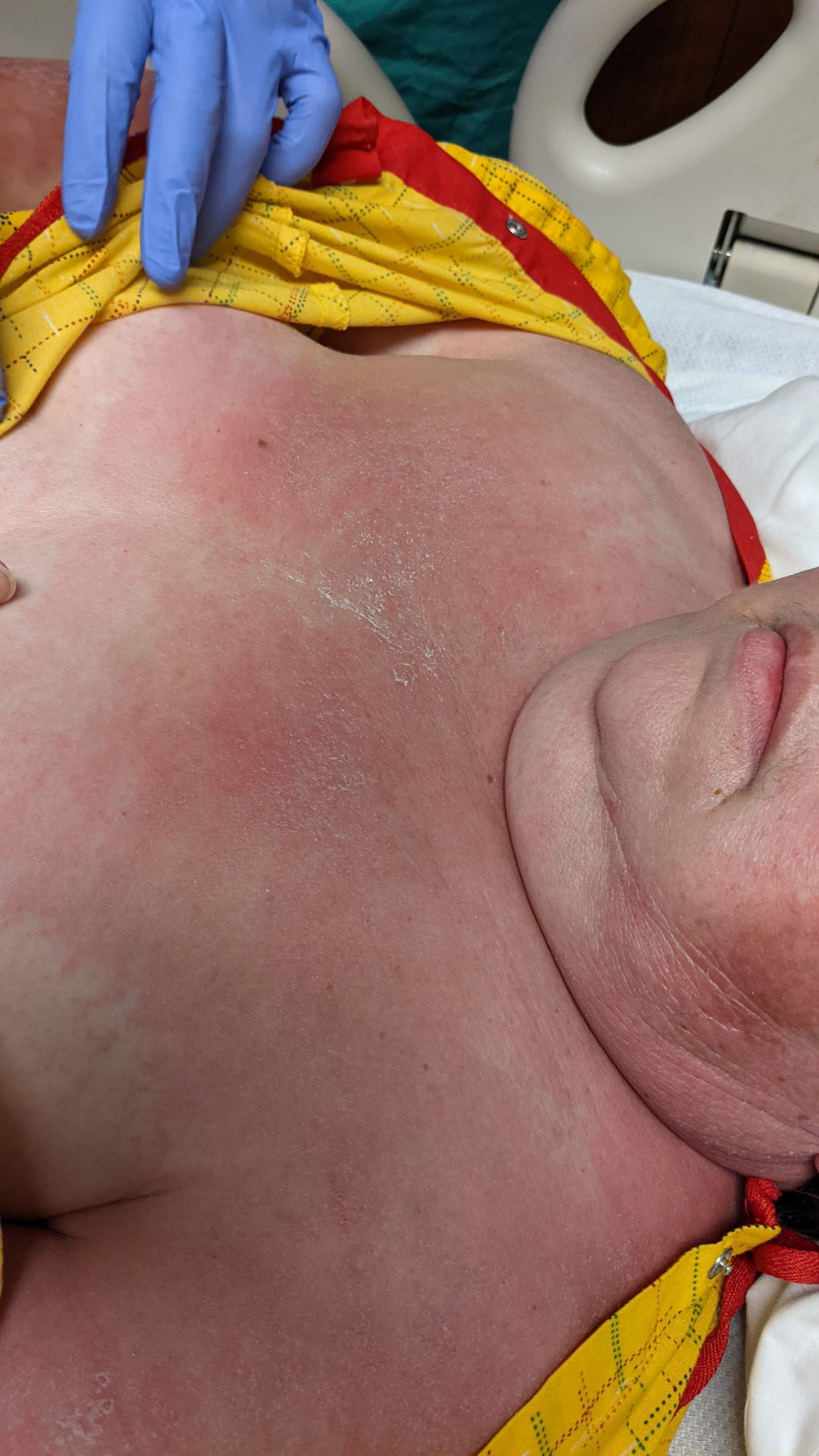
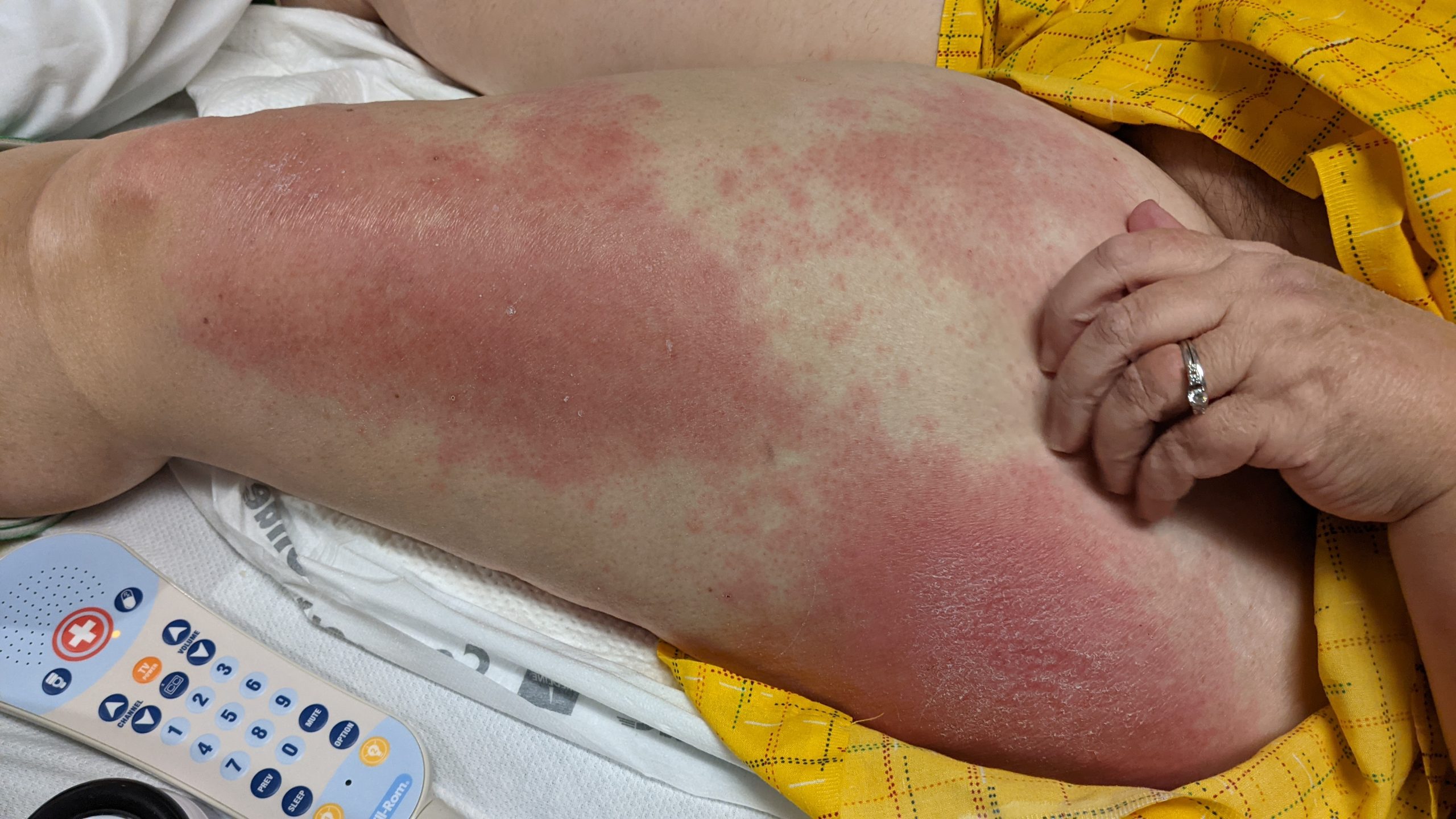
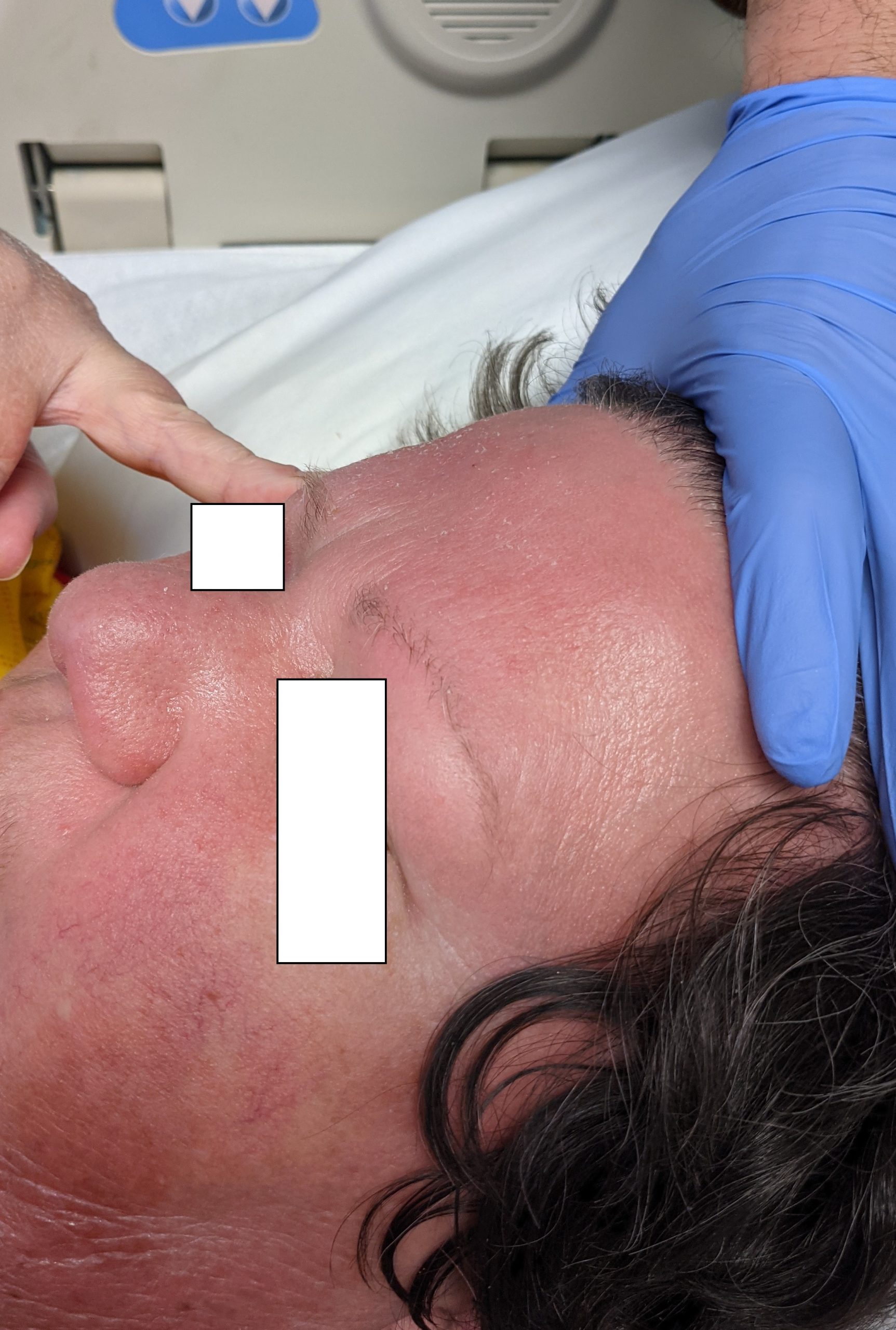
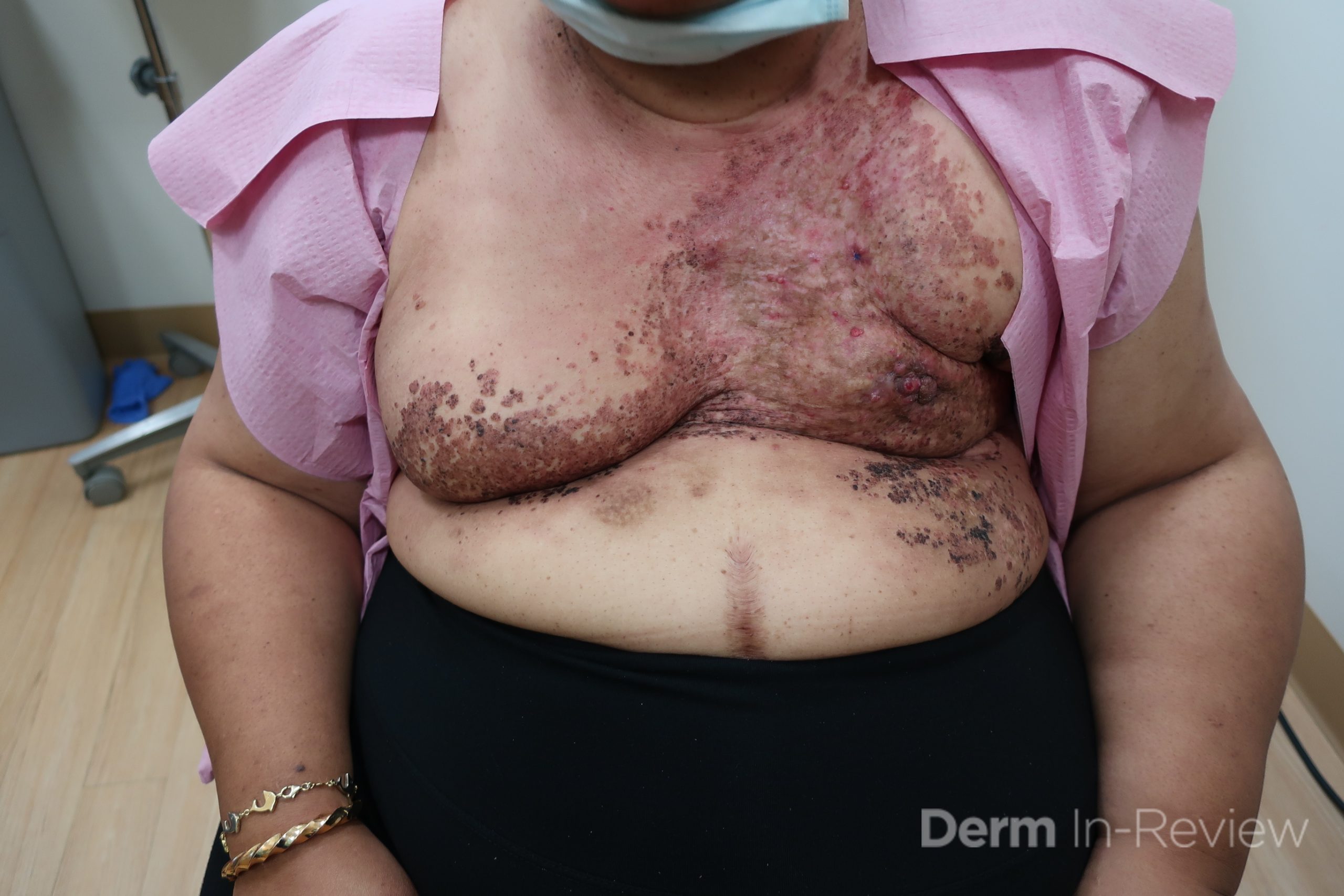
Patient is a 54 year old woman who presents with progressive rash on the anterior torso for 3 months, which she describes as slightly tender bumps. She has history of early stage breast cancer on the left breast treated with lumpectomy and radiation, both completed 9 years ago. She denies any other concerning symptoms.
Biopsy of a representative lesion is most likely to show which of the following?
A.) Dermis infiltrated by atypical neoplasm formed by pleomorphic epithelial cells with hyperchromatic nuclei and distinct nucleoli, disposed in columns and glandular-like structures, amid desmoplastic collagen without associated inflammation, positive immunostaining for HER2, estrogen receptor, and progesterone receptor
B.) Atypical hyperchromatic epithelial cells forming crack-like vascular spaces between collagen bundles, positive immunostaining for CD34, CD31
C.) New vessels wrapping around vascular and adnexal structures, positive immunostaining for HHV-8
D.) Dilation of ectatic superficial vessels, loss of adnexa, papillary dermal pallor, lack of lymphoid band
E.) Acanthosis, pseudoepitheliomatous hyperplasia with suprabasilar crypts of eosinophils
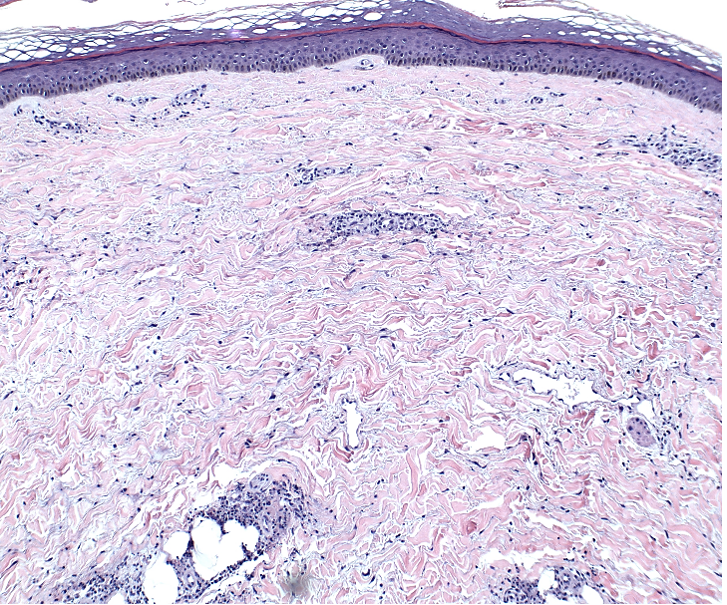
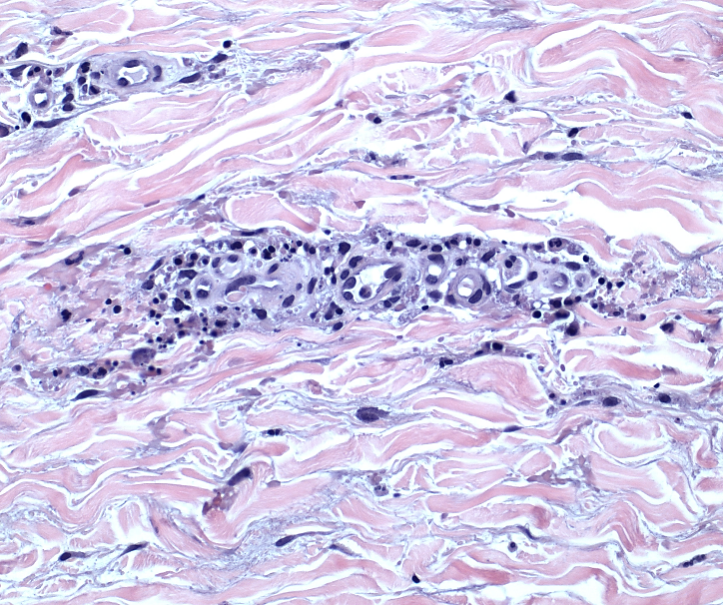
Correct answer: (A) Dermis infiltrated by atypical neoplasm formed by pleomorphic epithelial cells with hyperchromatic nuclei and distinct nucleoli, disposed in columns and glandular-like structures, amid desmoplastic collagen without associated inflammation, positive immunostaining for HER2, estrogen receptor, and progesterone receptor
Explanation:
This patient with history of early stage breast cancer who completed surgical and radiation treatment 9 years ago presents with pink to violaceous and hyperpigmented plaques, some studded with pink papules with overlying hemorrhagic crust, predominantly located on the left side of the chest and sprawling out toward the neck and the right chest. Her presentation is most consistent with carcinoma en cuirasse, a presentation of cutaneous metastases most often associated with breast cancer primary. Cutaneous metastases occur in 5-10% of cancer patients and may result from any malignant neoplasm. Prognosis associated with this diagnosis is very poor, as 6 and 12-month mortality rates for these patients are 48% and 64.5%, respectively. Presentations vary, with carcinoma en cuirasse characterized by a diffuse infiltration into the skin, leading to sclerodermoid change. Histopathology may be characterized by a (A) dermis infiltrated by atypical neoplasm formed by pleomorphic epithelial cells with hyperchromatic nuclei and distinct nucleoli, disposed in columns and glandular-like structures, amid desmoplastic collagen without associated inflammation. Positive immunostaining for HER2, estrogen receptor, and progesterone receptor would be suggestive of breast cancer primary. Other presentations of cutaneous metastases include: dermal or subcutaneous nodules, carcinoma erysipeloides, carcinoma telangiectodes, Paget’s disease, alopecia neoplastica, and pyogenic granuloma-like lesions.
Lymphangiosarcoma is high on the differential diagnosis for this patient’s presentation, and may present with non-healing eschars and multiple violaceous nodules. Stewart-Treves syndrome is a rare form of angiosarcoma which arises in the setting of chronic lymphedema, most commonly described in breast cancer patients who have undergone axillary lymph node dissection. Histopathology for this condition may show (B) atypical hyperchromatic epithelial cells forming crack-like vascular spaces between collagen bundles, with positive immunostaining for CD34, CD31. Patients usually have history of radical mastectomy, as opposed to lumpectomy, and lesions most commonly occur on the ipsilateral upper extremity overlying specific regions of lymphedema, as opposed to widespread lesions occurring over the torso.
Histopathology showing (C) new vessels wrapping around vascular and adnexal structures, with positive immunostaining for HHV-8 would be characteristic of Kaposi sarcoma (KS). There are 4 types of KS, which results directly from human herpesvirus-8 (HHV-8) infection. The classic type usually affects male descendants from the Mediterranean and middle European areas, as well as men in Sub-Saharan Africa, and is not associated with immunodeficiency. Human immunodeficiency virus (HIV)-associated KS is associated with severe immunodeficiency secondary to HIV infection. Endemic KS arises in some parts of Africa, usually in younger patients. Iatrogenic KS results from drug-induced immunosuppression. Presentation classically consists of red to violaceous macules, plaques, or nodules localized anywhere on the skin or mucous membranes, and may even involve the lymph nodes or viscera. There are numerous diverse morphologies reported in the literature, however, including generalized lymphadenopathic, exophytic, disseminated cutaneous and visceral, telangietatic, keloidal, ecchymotic, and localized nodular, plaque, or patch. In this patient who is otherwise well, has no history of immunosuppression, has no known ancestry from any of the aforementioned regions, these widespread tender lesions are more likely representative of cutaneous metastases.
Histopathology showing (D) dilation of ectatic superficial vessels, loss of adnexa, papillary dermal pallor, and lack of lymphoid band would be characteristic of chronic radiation dermatitis. Radiation dermatitis results from direct exposure to radiotherapy delivered by way of external beam ionizing radiation. Acute radiation dermatitis may present as erythema, desquamation, skin necrosis, and ulceration in areas of skin that have been irradiated. Chronic radiation dermatitis may occur up to 10 years or more after the beginning of radiotherapy and involves telangiectasia, fragile skin, and loss of follicular structures. In addition to the current patient’s distinctive morphology suggestive of cutaneous metastases, her radiation was 9 years ago and her eruption is very widespread, making radiation dermatitis much less likely.
Histopathology showing (E) acanthosis, pseudoepitheliomatous hyperplasia with suprabasilar crypts of eosinophils would be suggestive of pemphigus vegetans. Pemphigus vegetans is a localized form of pemphigus vulgaris. Early lesions may present as flaccid pustules, leading to erosions and vegetative and papillomatous plaques usually involving intertriginous areas, scalp, and face. There are 2 predominant subtypes, with the Hallopeau type consisting of more benign disease with longer periods of remission, and the Neumann type involving more disease relapses and requiring higher doses of corticosteroids. Similar to other forms of pemphigus vulgaris, patients have autoantibodies to desmoglein 3 predominantly. This current patient’s lesion morphology and areas of involvement make pemphigus vegetans an unlikely diagnosis.
References
- Bolognia JL, Schaffer JV, Duncan KO, Ko CJ. Dermatology essentials E-book. Elsevier Health Sciences; 2014 Feb 26.
- Elston D, Ferringer T, Ko CJ, Peckham S, High WA, DiCaudo DJ. Dermatopathology E-Book. Elsevier Health Sciences; 2013 Oct 16.
- Schoenlaub, P., et al. “Survival after cutaneous metastasis: a study of 200 cases.” Annales de Dermatologie et de Venereologie. Vol. 128. No. 12. 2001.
- Oliveira GM, Zachetti DB, Barros HR, Tiengo A, Romiti N. Breast carcinoma en Cuirasse-case report. Anais brasileiros de dermatologia. 2013 Aug;88(4):608-10.
- Mahore SD, Bothale KA, Patrikar AD, Joshi AM. Carcinoma en cuirasse: A rare presentation of breast cancer. Indian Journal of Pathology and Microbiology. 2010 Apr 1;53(2):351.
- Sharma A, Schwartz RA. Stewart-Treves syndrome: pathogenesis and management. Journal of the American Academy of Dermatology. 2012 Dec 1;67(6):1342-8.
- Etemad SA, Dewan AK. Kaposi sarcoma updates. Dermatologic clinics. 2019 Oct 1;37(4):505-17.
- Hegedus F, Mathew LM, Schwartz RA. Radiation dermatitis: an overview. International journal of dermatology. 2017 Sep;56(9):909-14.
- Razzaque Ahmed A, Blose DA. Pemphigus vegetans: Neumann type and Hallopeau type. International journal of dermatology. 1984 Mar;23(2):135-41.