Correct Answer: B.) Erythromycin
A) Incorrect. Griseofulvin is an antifungal agent that prevents dermatophyte mitosis via tubulin inhibition. It is primarily utilized for pediatric patients to treat tinea capitis caused by Microsporum canis.1
B) Correct. The clinical and histopathologic findings for this case are consistent with bacillary angiomatosis. Erythromycin, a macrolide antibiotic, is the first line therapy for treatment.2 Macrolides inhibit bacterial synthesis by binding to the 50s ribosomal subunit.1 Additionally, they are well known CYP3A4 inhibitors, which can increase risk for torsade de pointes when combined with other medications.2 Thus, it is important to review patient medications prior to starting macrolides.
C) Incorrect. Cephalexin is a first generation cephalosporin that treats streptococcal and staphylococcal infections such as uncomplicated cellulitis, impetigo, or furuncolosis.2 Common side effects include GI upset and hypersensitivity reactions. About 5-10% of penicillin-allergic patients can experience crossreactivity with cephalosporin use.1
D) Incorrect. Ciprofloxacin is a first generation fluoroquinolone with gram-negative coverage.1 This class of antibiotics interferes with DNA synthesis via inhibition of DNA gyrase and topoisomerase IV.1 Side effects of fluoroquinolones include GI upset, tendinitis, tendon rupture, and photosensitivity.1 Ciprofloxacin can be used to treat Oroya fever rather than bacillary angiomatosis.2
E) Incorrect. Valacyclovir is an antiviral agent used to treat herpes simplex infections and varicella-zoster infections. It is a prodrug of acyclovir with increased bioavailability.1 Valacyclovir is generally well tolerated with minimal side effects. Rarely, immunosuppressed patients can experience thrombotic thrombocytopenic purpura or hemolytic uremic syndrome with use of valacylovir.2
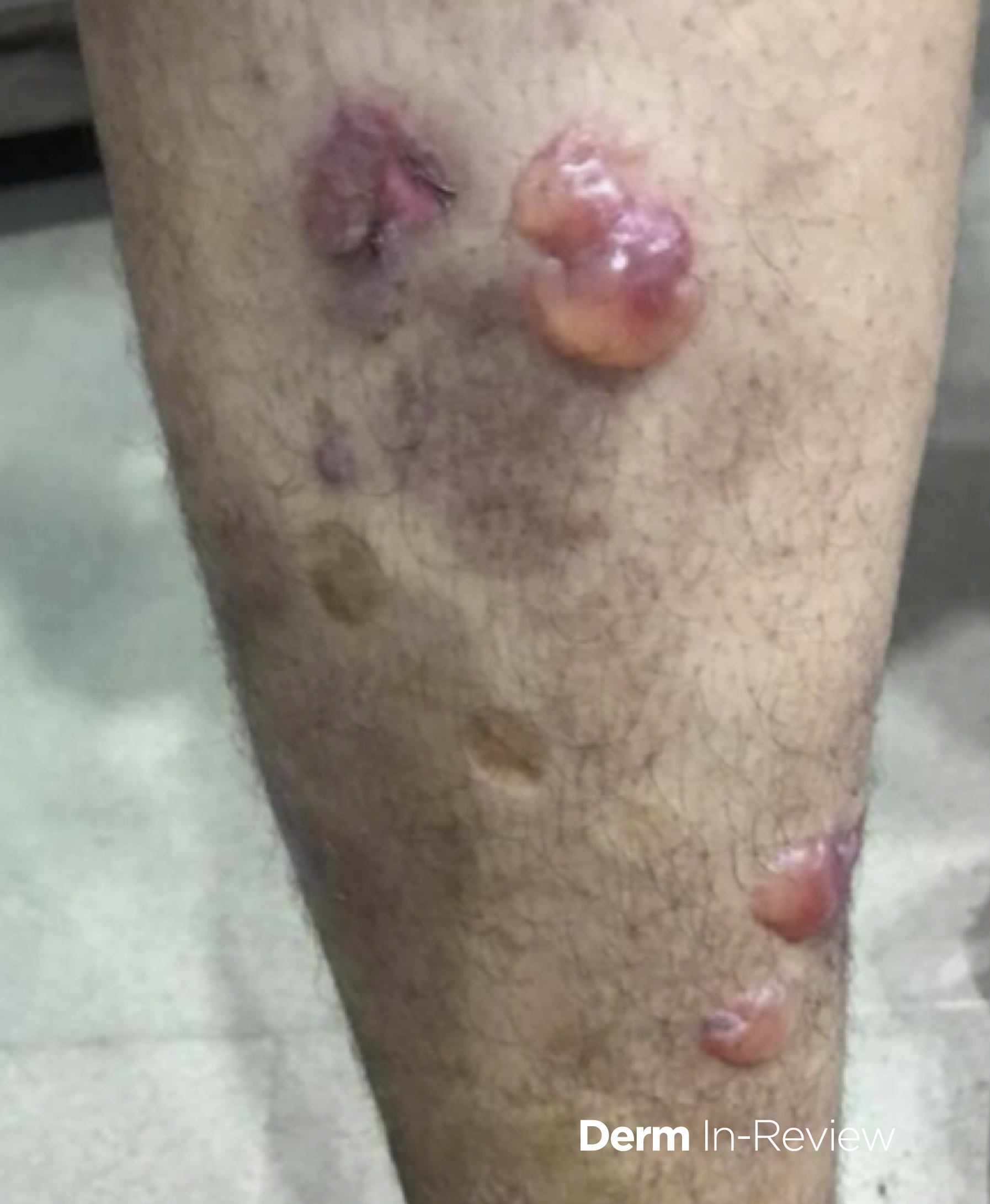
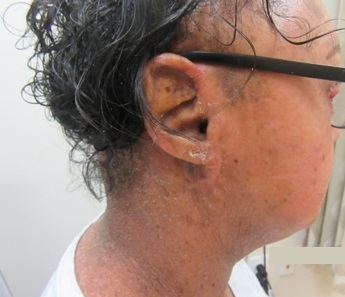
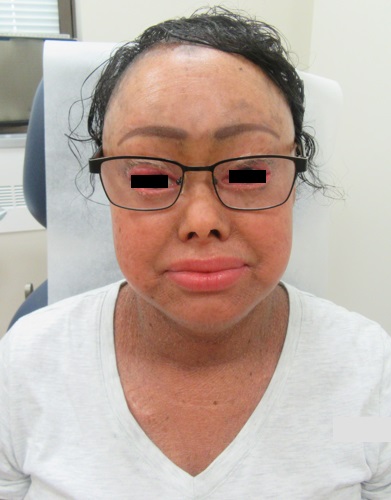
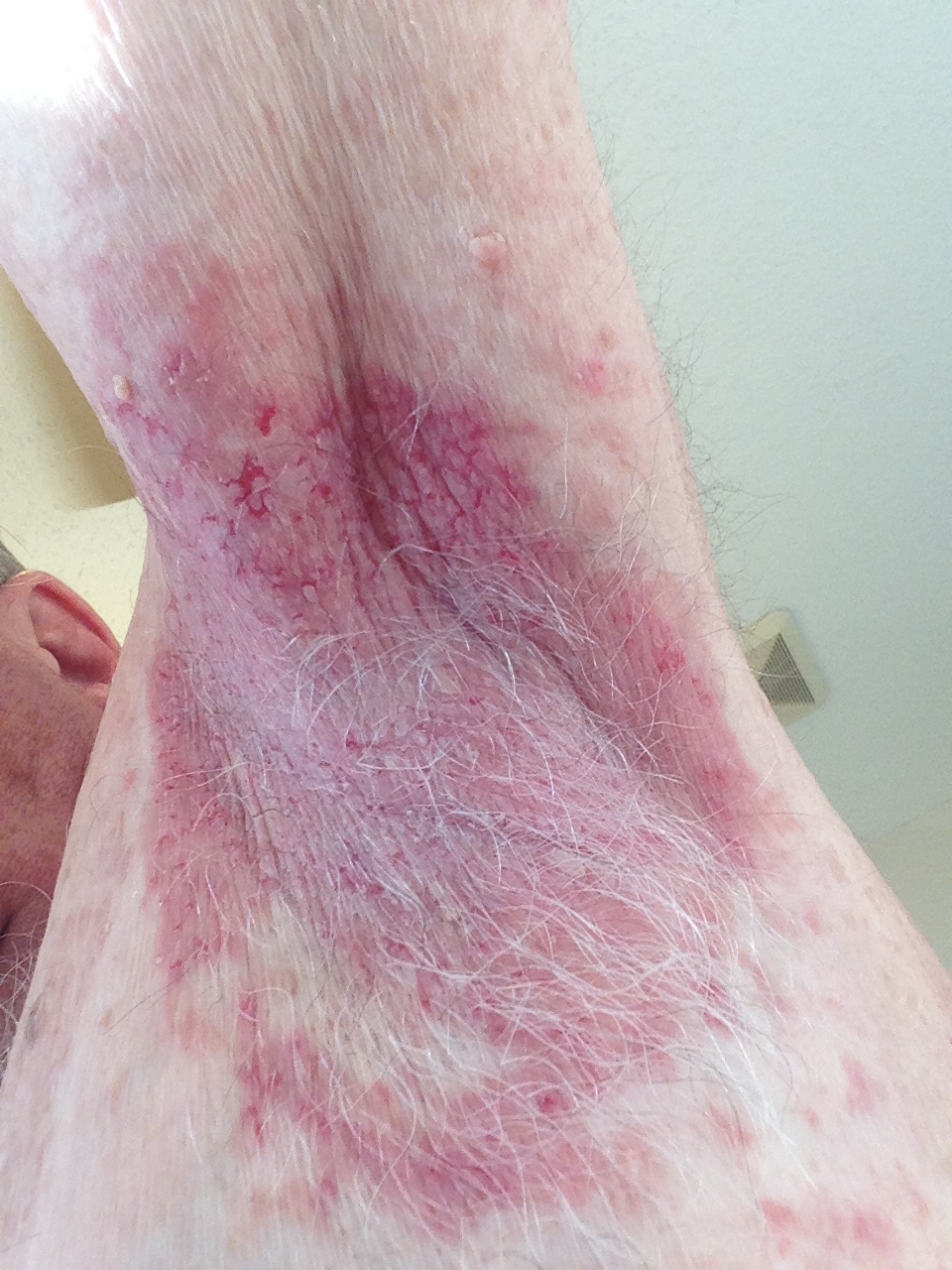
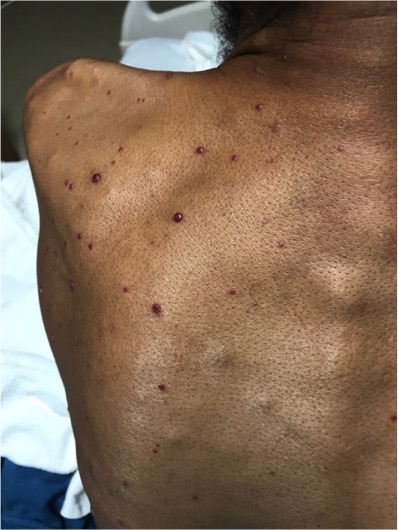
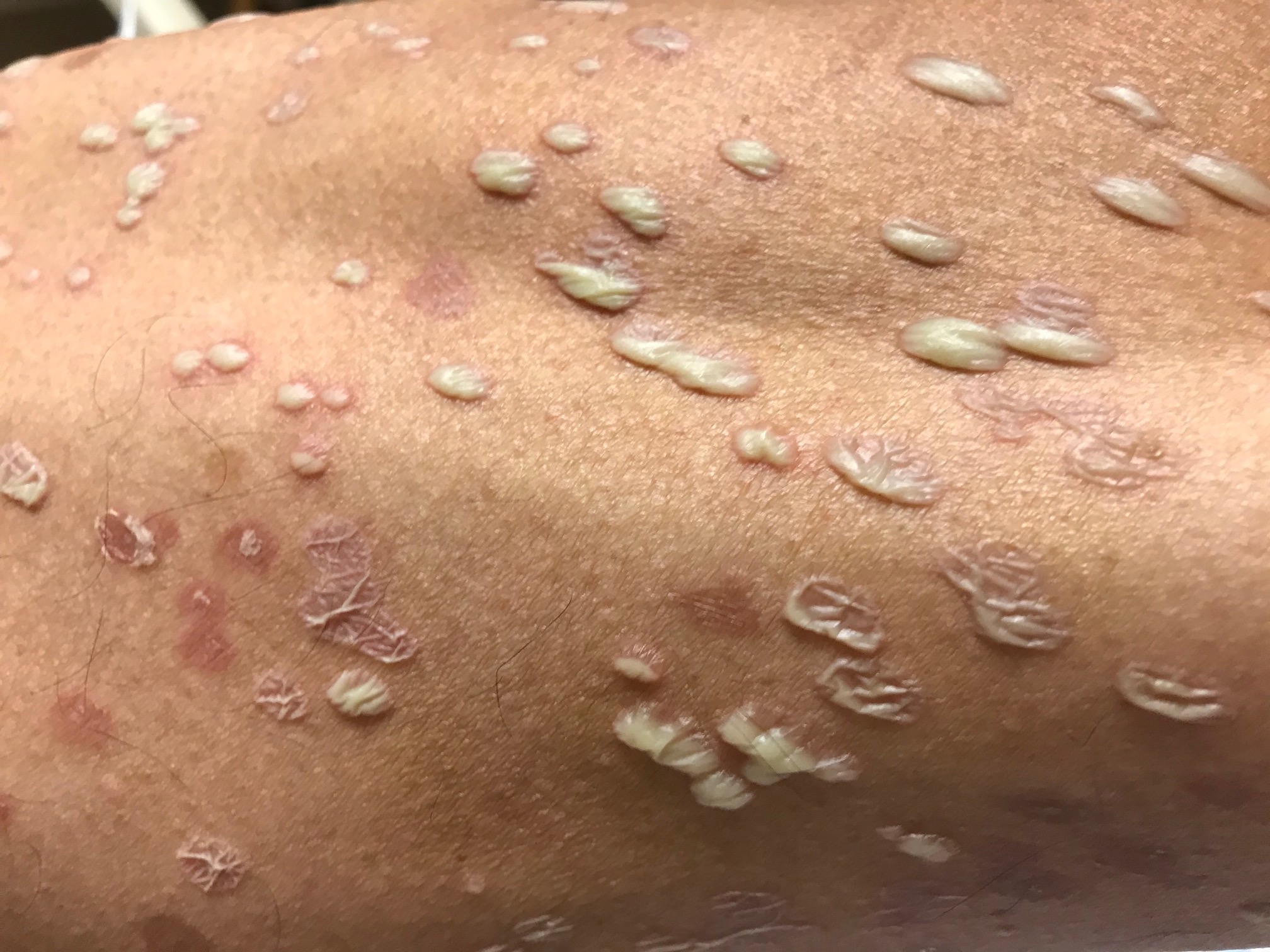
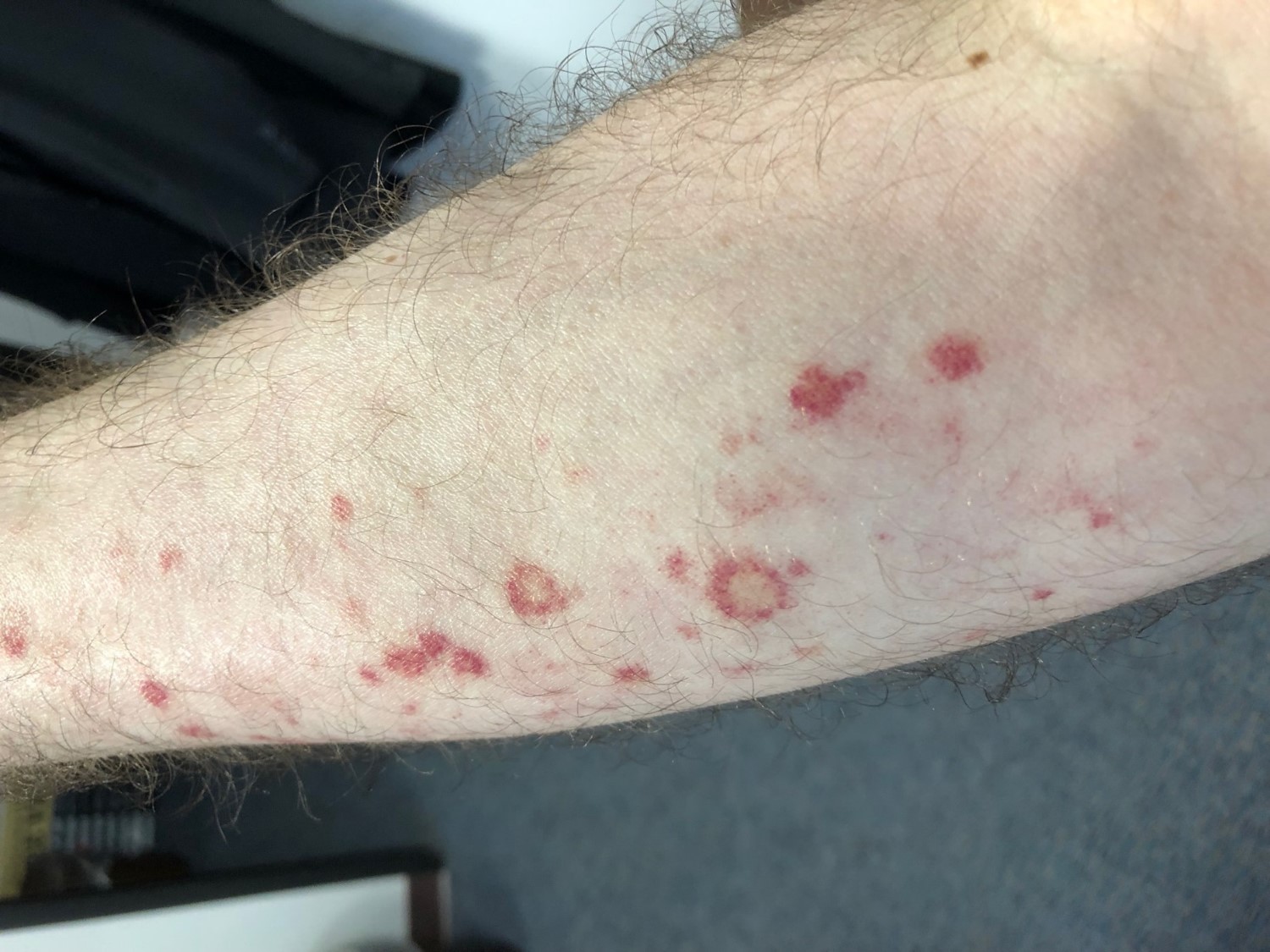
Bacillary angiomatosis is one of the several conditions caused by Bartonella infections. There are over thirty species of these gram-negative bacilli, but only three species are known to affect humans – B. henslae, B. quintana, B bacilliformis.2 Other diseases associated with Bartonella infections include cat scratch disease, Oroya fever, verruga peruana, Carrion disease, endocarditis, and bacillary peliosis.2
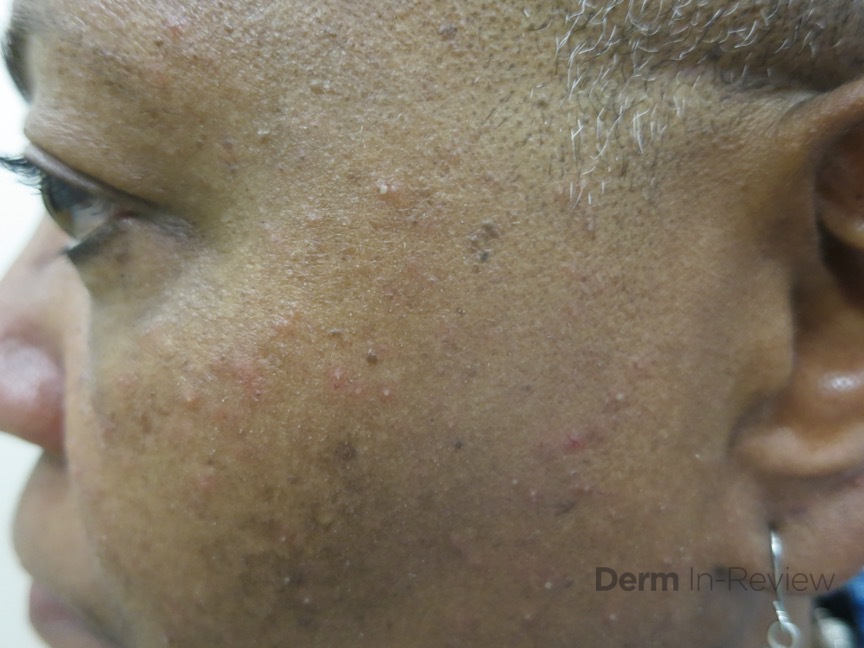
Bacillary angiomatosis is commonly seen in immunosuppressed states, such as HIV and organ transplant recipients, and is caused by B. henslae and B. quintana. Vectors for these Bartonella species are the cat flea and human body louse, respectively.2
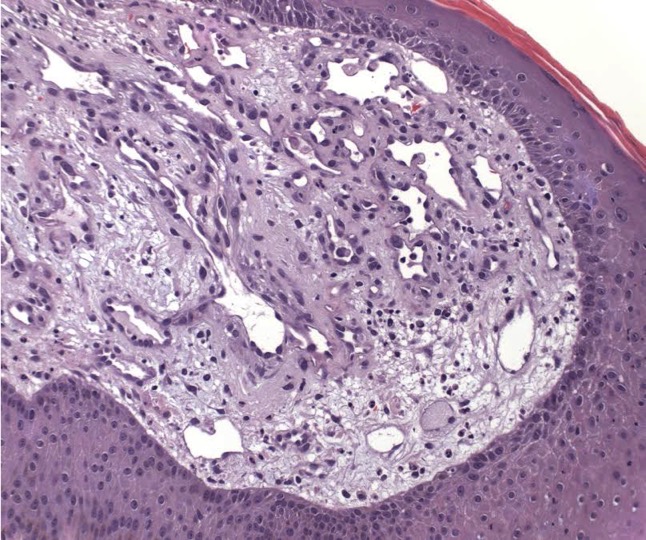
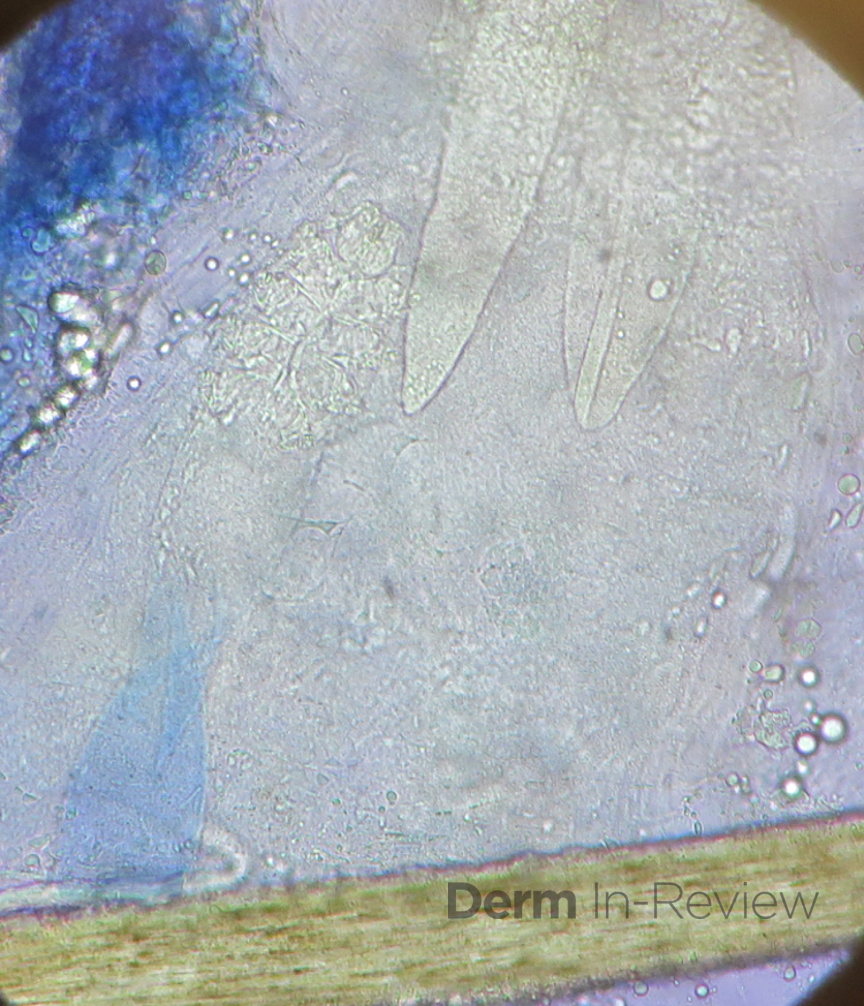
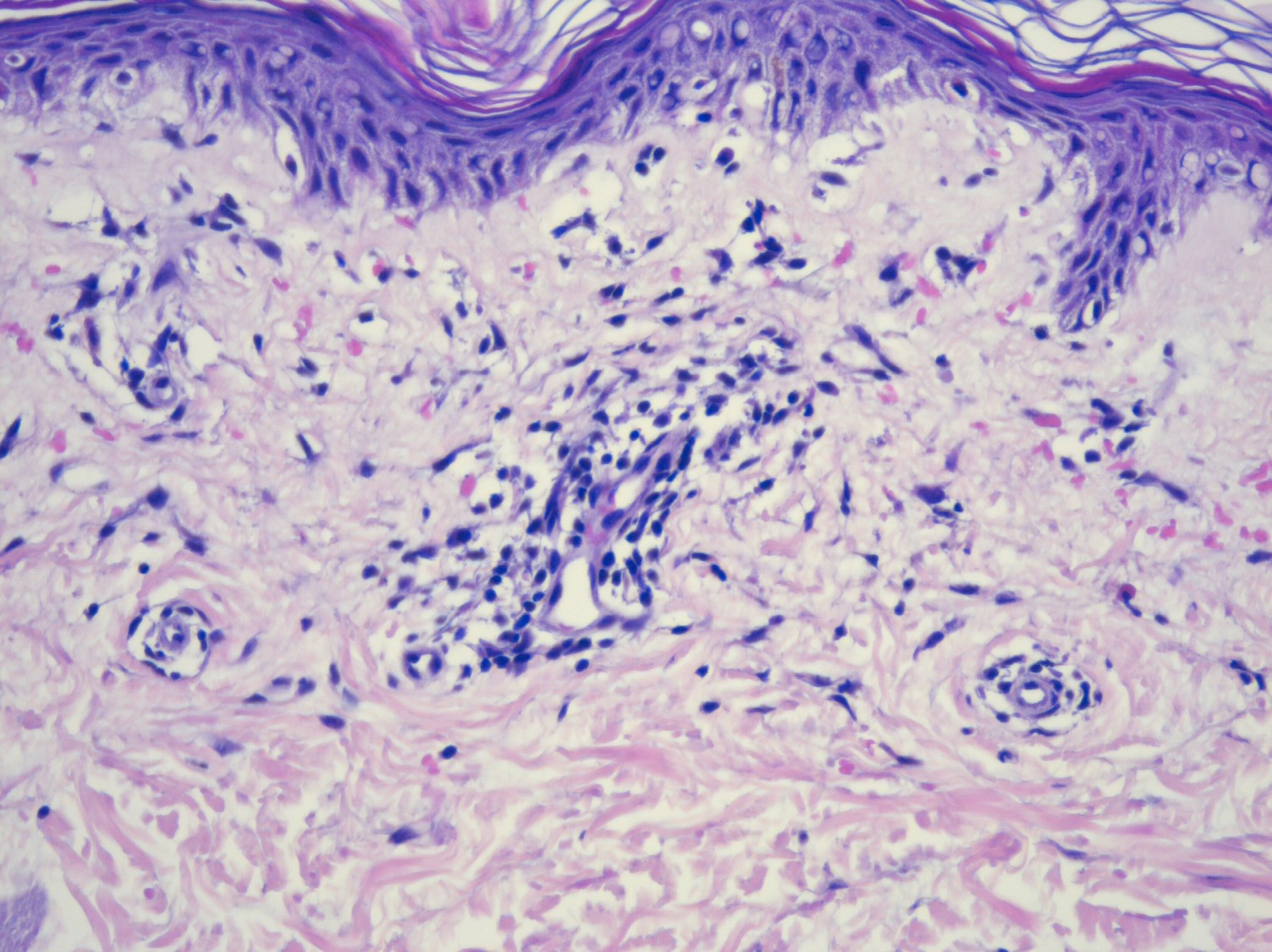
There should be a high suspicion for bacillary angiomatosis in the correct clinical scenario and histopathologic findings can often confirm diagnosis. It is important to differentiate bacillary angiomatosis from Kaposi sarcoma given histopathology similarity.2 This can be achieved with concomitant HHV-8 and Warthin-Starry staining. Unfortunately, Bartonella bacilli are not always identified on Warthin-Starry staining. Thus, serologic testing for B. henslae and B. quintana immunoglobulins or serum DNA PCR should also be considered.
First line treatment for bacillary angiomatosis erythromycin dosed at 500mg every six hours.2 However, doxycycline 100mg every twelve hours is an appropriate, and sometimes preferred, alternative due to less frequent dosing. Additional treatment options include azithromycin and clarithromycin.2 In severe cases of bacillary angiomatosis, combination therapy with doxycycline and gentamicin, doxycycline and rifampin, or azithromycin and gentamicin should be utilized.2
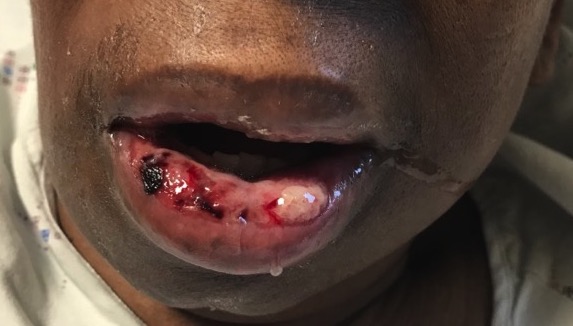
While bacillary angiomatosis is classically associated with immunosuppression, this infection can also occur in immunocompetent patients. Several cases of bacillary angiomatosis arising within second degree burns of immunocompetent patients have been reported.3,4 Both patients had complete resolution with oral erythromycin.3,4