December 2020 Case Study
by Kamaria Nelson, MD, MHS
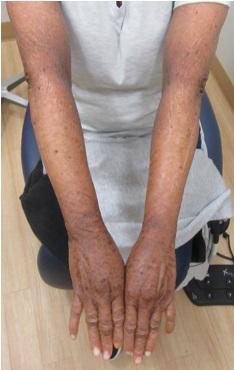
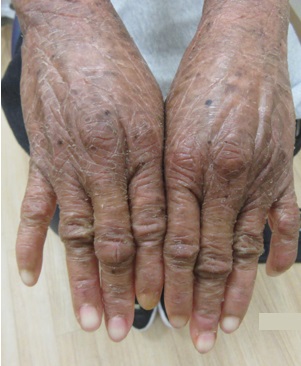
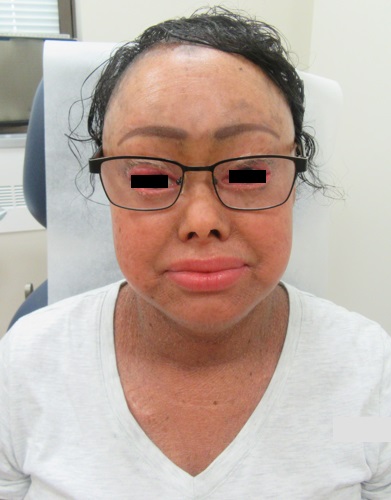
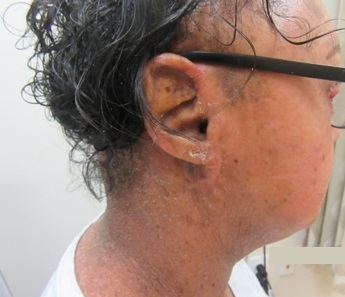
A 33-year-old female presents with a rash that she describes as “thick, excess skin.” The rash has been present since birth. She reports difficulty with regulating her temperature and having dry eyes due to her condition. Current treatments include urea, petroleum jelly, and n-acetyl cysteine. She was on oral acitretin from ages 3-14. The patient reports no other history of cutaneous disease or systemic conditions. She has a sister with similar skin findings. Physical examination reveals thick, plate-like, dark appearing scaly plaques on the arms, upper chest, neck and face; ectropion and dysmorphic appearing facies were present. (Images 1-4).
Based upon the patient history and clinical examination, which genetic mutation is most likely related to this condition?
A.) ALDH3A2 (or ALDH10)
B.) TGM1
C.) Genes encoding KRT1 & KRT10
D.) ABCA12
E.) STS
Correct answer: B) TGMI
Lamellar ichthyosis (LI) in an autosomal recessive congenital ichthyosis characterized by diffuse large, gray brown scales that are quadrilateral and can resemble an armor plate. During the first 2-3 weeks of life, LI frequently presents with a collodion membrane with ectropion that then desquamates. 35-55% of all patients have transglutaminase-1 deficiency due to mutations in TGM1. Transglutaminase-1 is found in the upper layers of the epidermis where it catalyzes the linking of proteins and aids in the formation of ε-lysine isopeptide bonds and ester bonds between proteins and ω-hydroxyceramides.
Transglutaminase-1 crosslinks structural proteins to each other to form the cornified cell envelope. The formation of the cornified cell envelope is important for intracellular lipid layer formation in the stratum corneum; therefore, mutations can cause defective barrier function. Mutations in CYP4F22 results in an acral, milder presentation of lamellar ichthyosis. CYP4F22 encodes an enzyme that’s responsible for ω-hydroxylation of ultra-long-chain fatty acids which form acyleramides, important to skin barrier function. Histologic features are not diagnostic and may reveal orthokeratotic hyperkeratosis that covers an acanthotic epidermis, normal epidermal proliferation rate, and a thin or absent cornified cell envelope on electron microscopy. Treatment consists of adequate moisturization with emollients. Topical retinoids can be used along with ammonium lactate and urea for adherent scale. For more severe cases, systemic retinoids, such as isotretinoin or acitretin, should be considered.
Sjogren-Larsson syndrome (SLS) is an autosomal recessive neurocutaneous disorder that is caused by mutations in ALDH3A2, which leads to a deficiency of fatty aldehyde dehydrogenase (FALDH). FALDH catalyzes oxidation of long-chain aliphatic aldehydes into fatty acids; mutations lead to accumulation of fatty alcohol/aldehyde-modified lipids and proteins causing membrane alteration. SLS is characterized by congenital ichthyosis, spastic paralysis, intellectual disability, and degenerative retinitis. Patients present with a juvenile macular dystrophy exhibited by perifoveal glistening white dots in the ocular fundus which are present during the first year of life. SLS is also associated with persistent pruritus, unlike the other ichthyoses.
Epidermolytic ichthyosis (EI), also known as epidermolytic hyperkeratosis, is an autosomal dominant disorder that is caused by heterozygous mutations in the genes that code for keratin 1 (KRT1) and keratin 10 (KRT10). KRT1 and KRT10 are expressed in the differentiated spinous and granular layers of the epidermis; mutations compromise epidermal integrity leading to cytolysis and blistering. EI usually presents with blisters at or shortly after birth and later, thick, uniquely malodorous spine-like scale on the flexures. Key histological findings include orthokeratotic hyperkeratosis, prominent acanthosis, hypergranulosis and cytolysis of suprabasal and granular layers; keratinocytes reveal intracellular vacuolization and dense clumps of keratin intermediate filaments (KIFs).
Harlequin ichthyosis (HI) is rare and the most severe form of autosomal recessive congenital ichthyosis. It is caused by loss-of-function mutations in ABCA12. ABCA12 is responsible for ceramide and lipid transport into lamellar bodies; mutations prevent the formation of lipid bilayers in the stratum corneum resulting in hyperkeratosis and a severe breakdown of the barrier. HI presents with thick collodion, diffuse armor-like plates and severe ectropion and eclabium. Patients with HI are usually born premature and have a high mortality rate (nearing 50%) due to respiratory insufficiency from severe immobilization caused by the thickened stratum corneum or sepsis from severely fissured skin.
X-linked ichthyosis results from a deficiency of steroid sulfatase caused by the deletion of the STS gene on chromosome Xp22.31. This results in inadequate hydrolysis of cholesterol sulfate and dehydroepiandrosterone sulfate (DHEAS) causing accumulation of cholesterol 3-sulfate in the epidermis which can inhibit tranglutaminase-1. Women who are pregnant with an affected fetus often have failure to initiate labor due to inadequate conjugation of DHEAS, necessary for estrogen synthesis. At birth, males may present with erythroderma and exfoliation of large, translucent scale. This later progresses to polygonal, dark-brown plates on the neck, extremities and trunk; periauricular involvement is characteristic. Other important findings include asymptomatic corneal opacities, increased incidence of cryptorchidism and risk for testicular cancer, and higher prevalence of attention deficit disorder. Molecular analysis (genome microarrays, FISH, PCR) can detect the genetic defect and may be used prenatally from amniotic samples. Noninvasive techniques include serum estriol levels and urine non-hydrolyzed sulfated steroids or biochemical assay to measure steroid sulfatase enzyme activity. Plasma cholesterol sulfate levels after birth can be used to aid in diagnosis.
References
- Bolognia, J., Schaffer, J., & Cerroni, L. (2019). Dermatology (Fourth edition.). Elsevier.
- James, W., Elston, D., Treat, J., Rosenbach, M., Neuhaus, I., & Andrews, G. (2013). Andrews’ Diseases of the Skin : Clinical Dermatology (Thirteenth edition.). Elsevier.