December 2023 Case Study
Adam Rosenfeld, MD1
- Department of Dermatology, George Washington University School of Medicine and Health Sciences
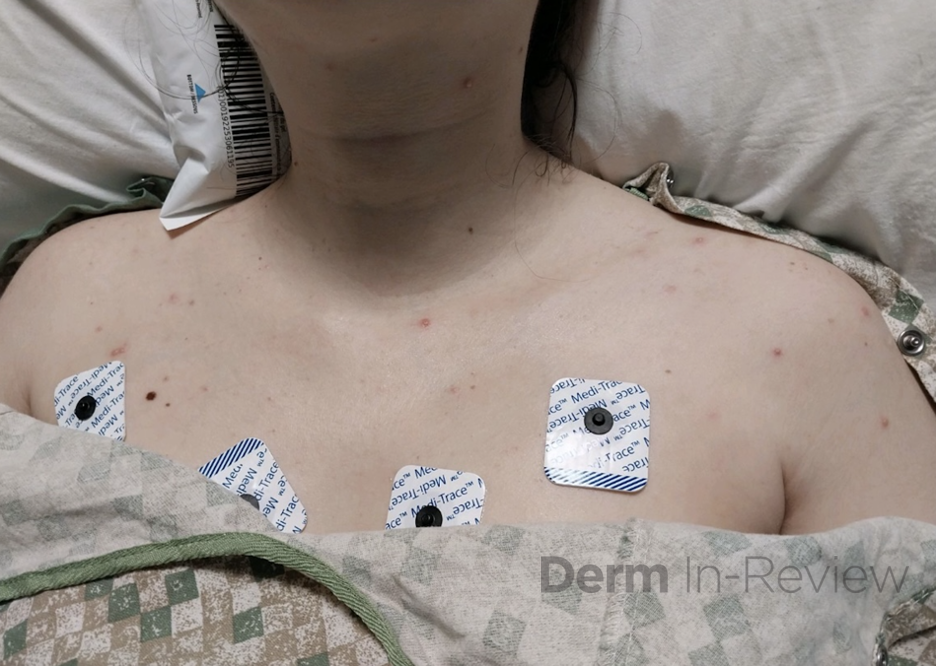
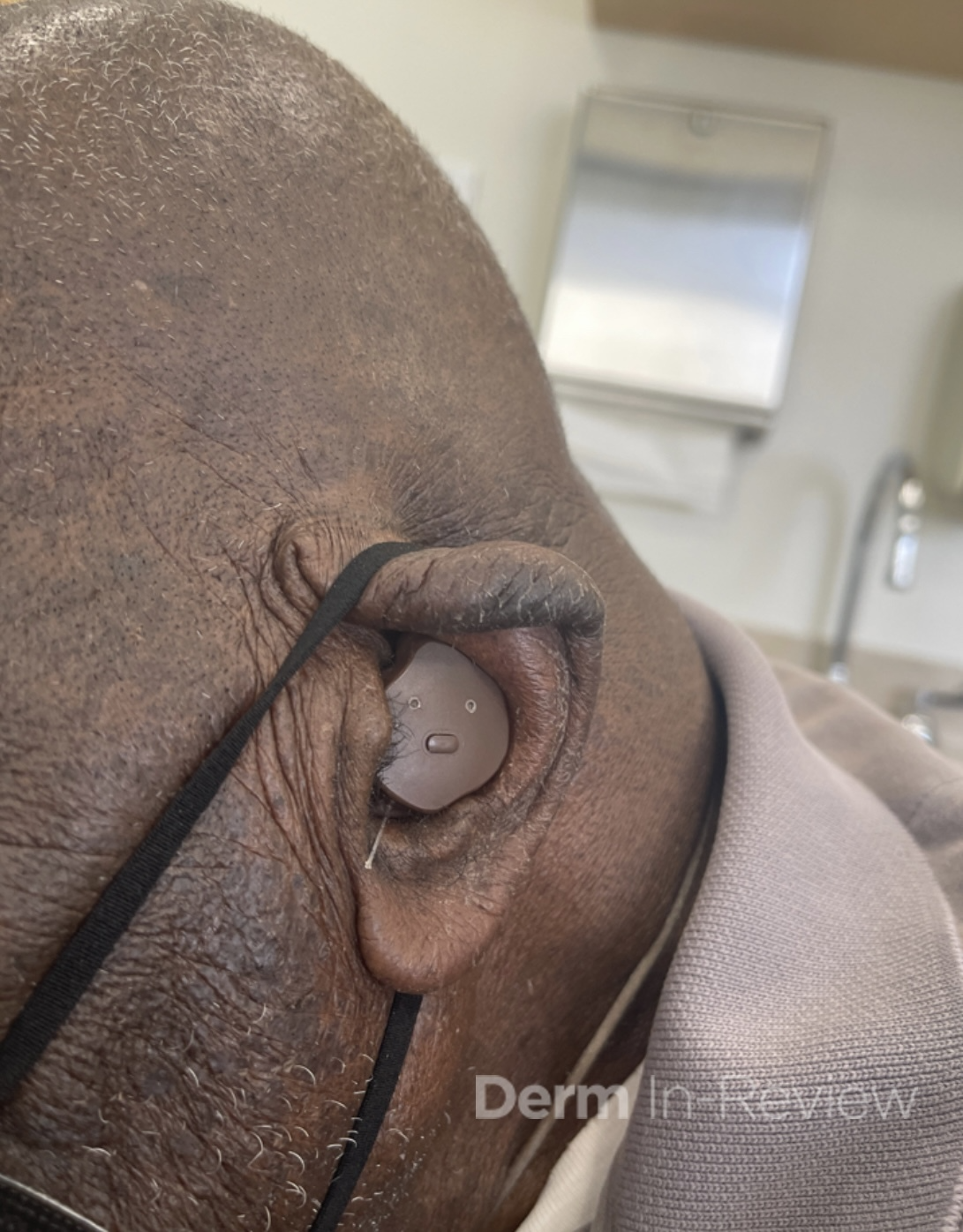
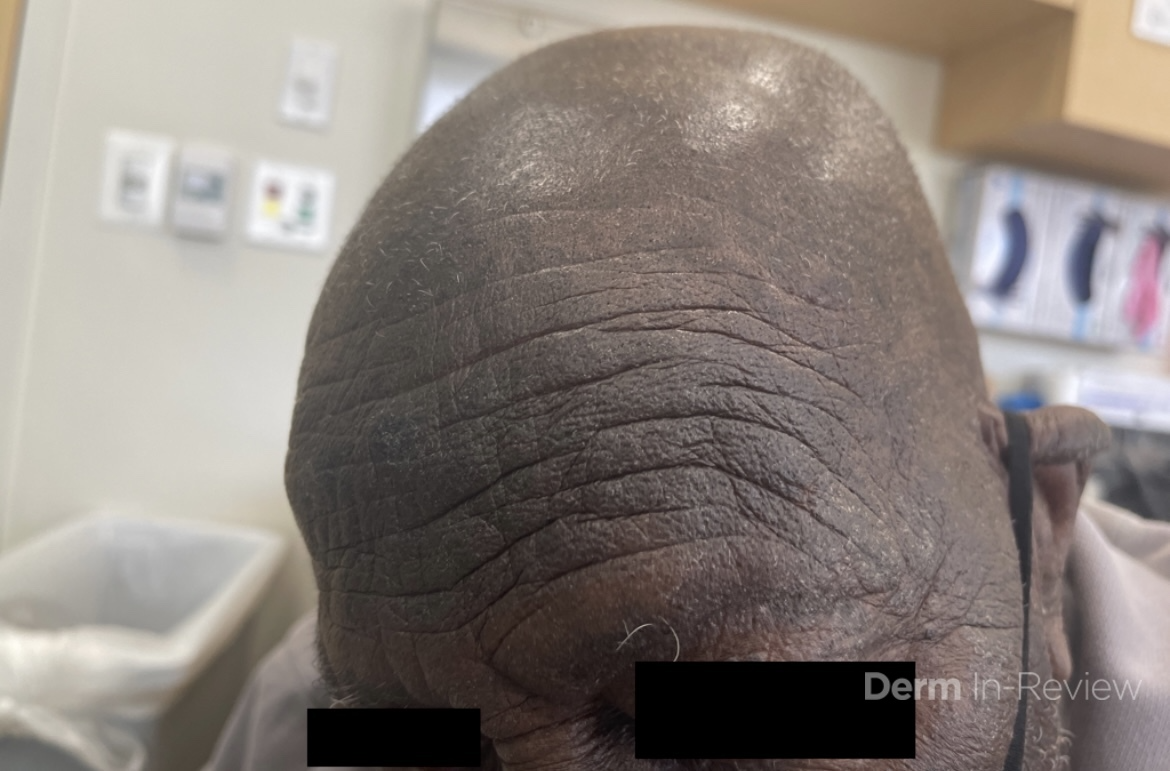
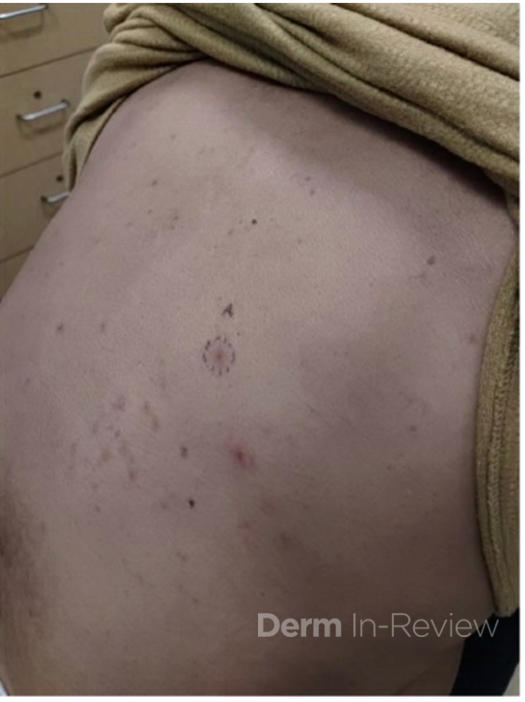
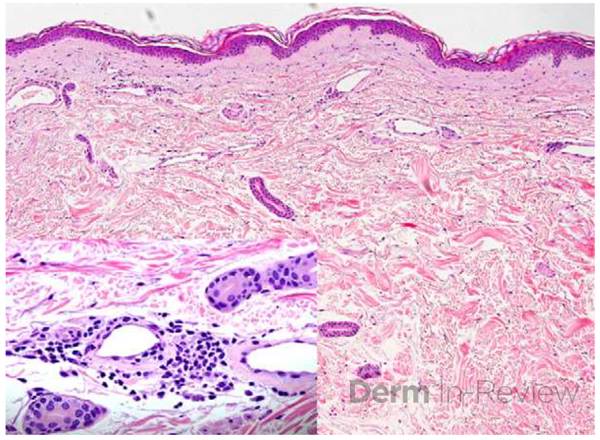
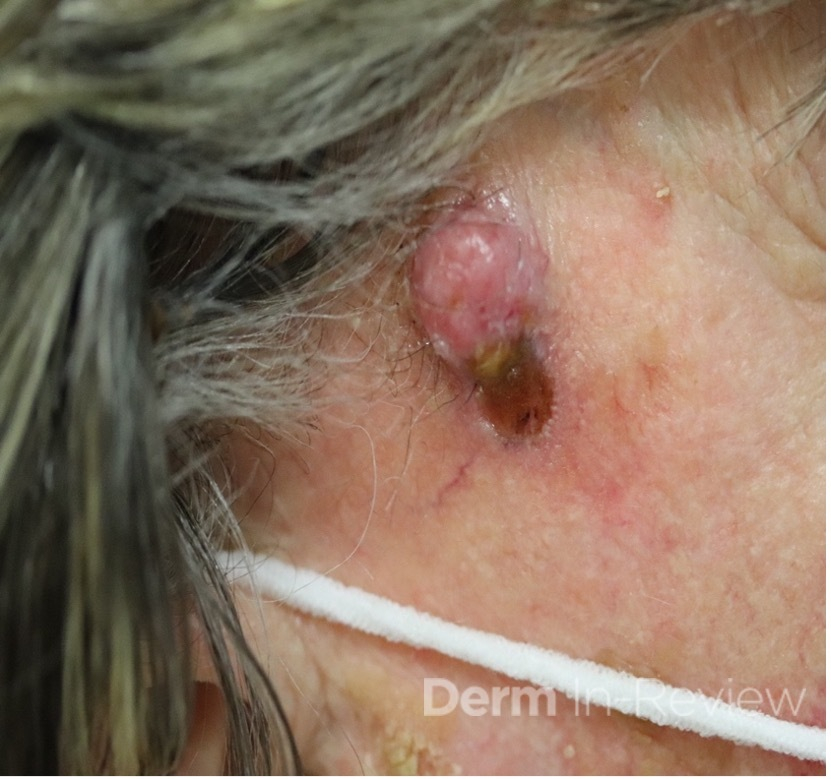
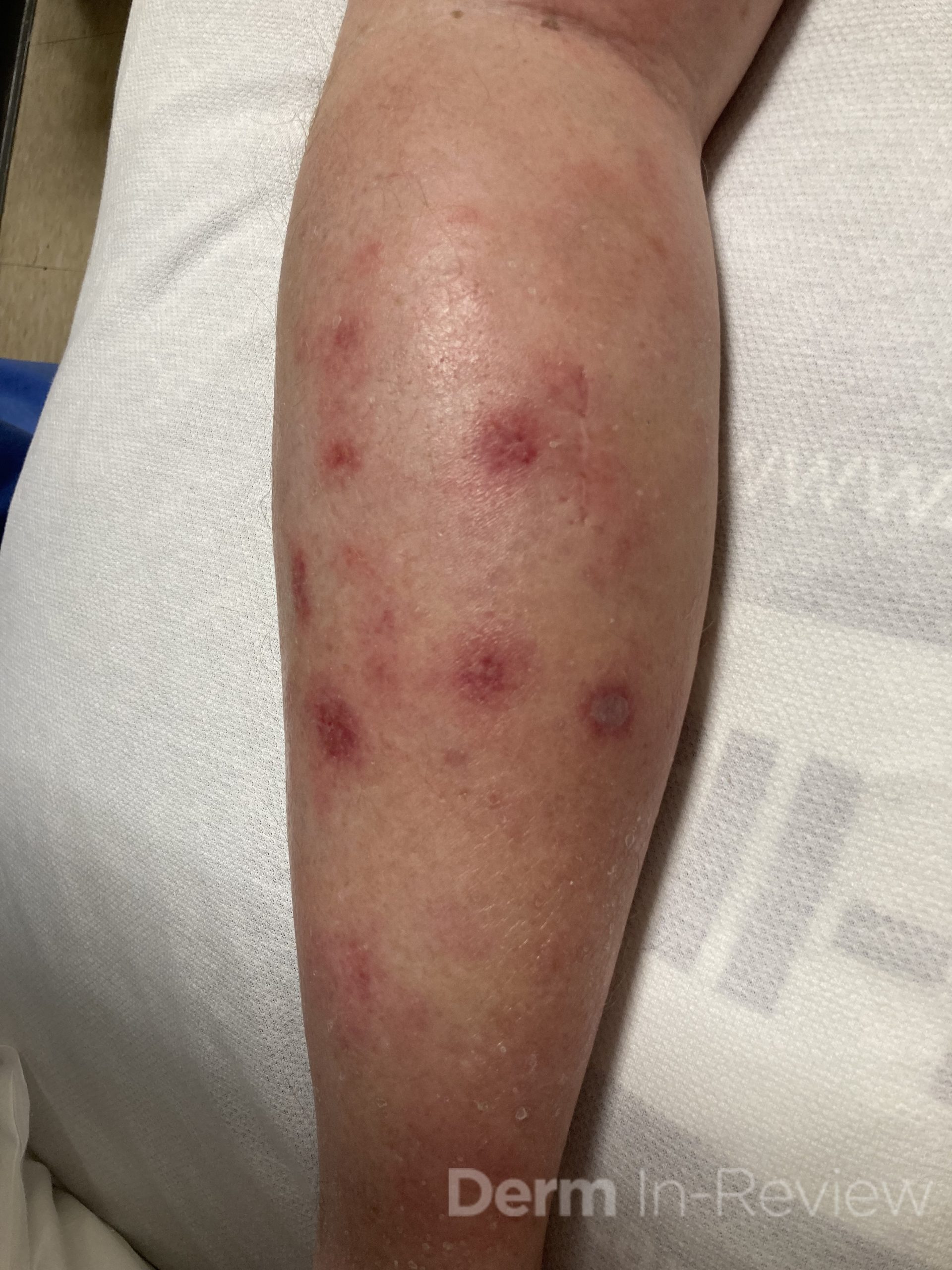
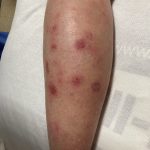
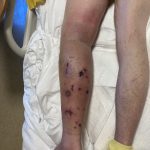
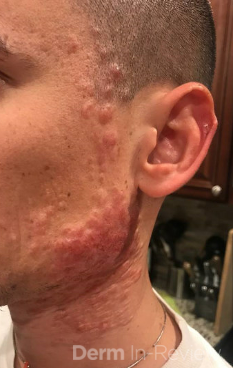
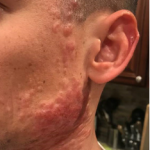
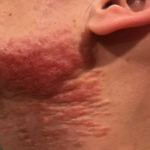
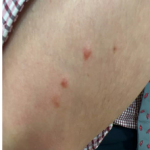
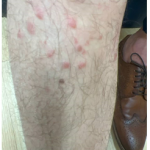
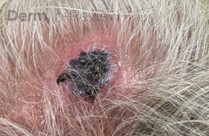
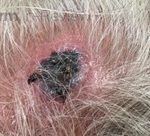
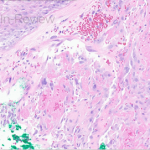
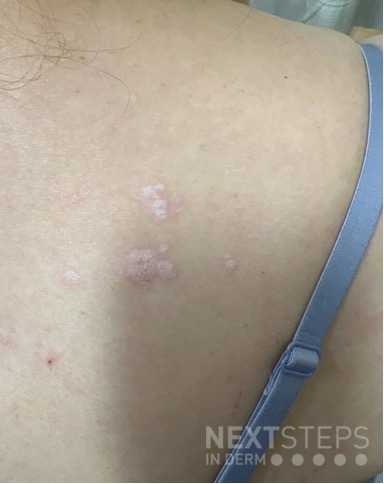
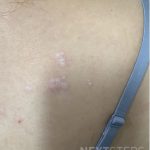
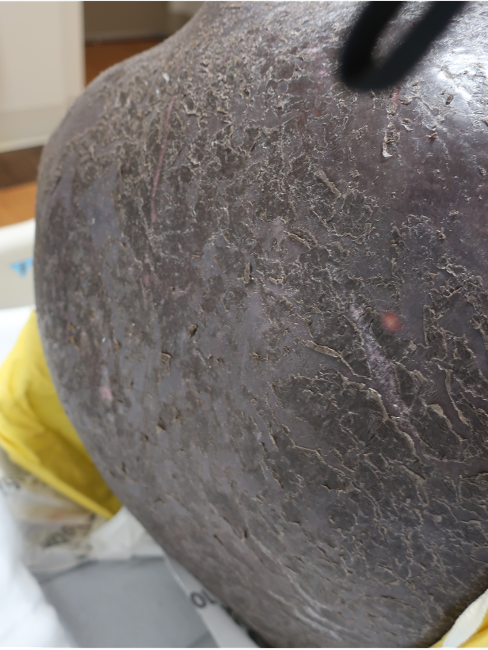
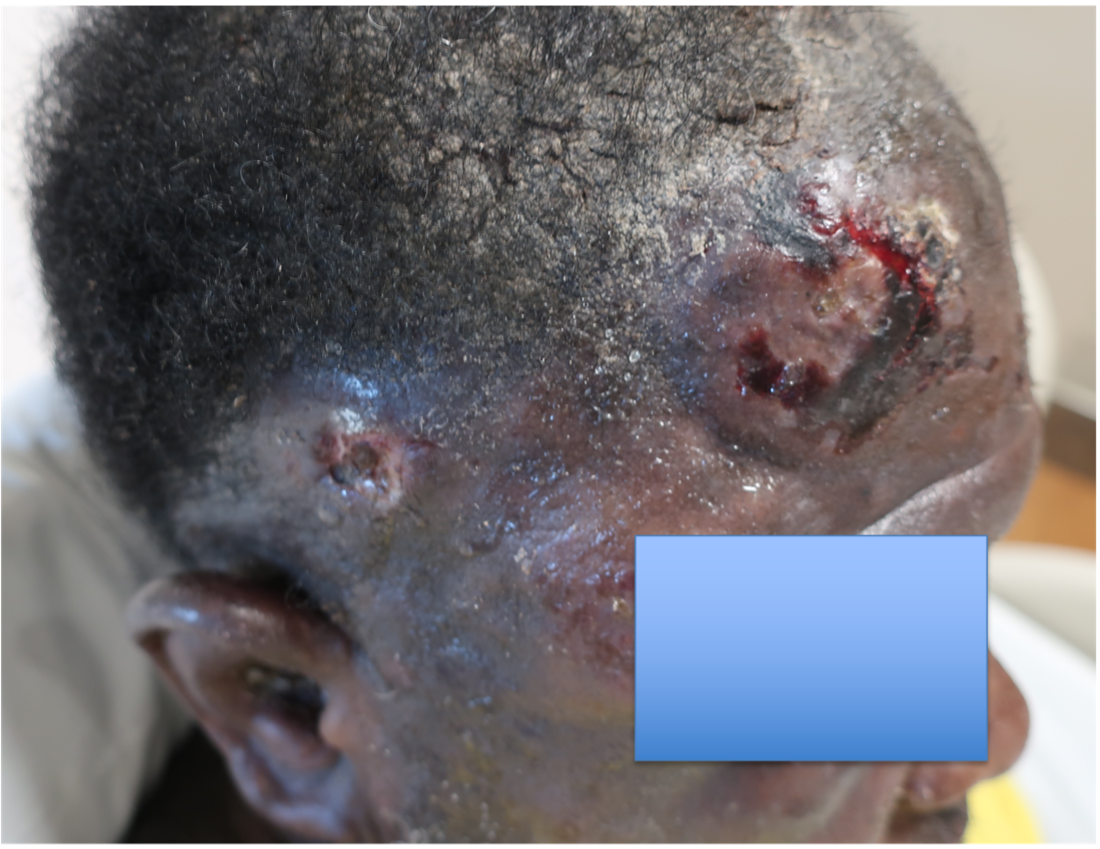
A 46-year-old male with reported history of atopic dermatitis presented to the ED with a 2–3 month history of a diffuse skin eruption as well as multiple growths on his forehead. Review of systems was notable for fatigue and chills. Patient endorsed unintentional weight loss and multiple episodes of night sweats throughout this time frame. On examination, the patient was erythrodermic with diffuse hyperpigmented and erythematous scaly plaques as well as multifocal, somewhat necrotic appearing nodules on the right forehead/temple area (figures 1,2). A punch biopsy demonstrated a superficial to deep dermal infiltrate of atypical lymphoid cells with limited epidermal involvement.
Given the overall presentation, loss of what T-cell marker would be most consistent with the likely diagnosis?
A.) CD20
B.) CD3
C.) CD4
D.) CD7
E.) CD45
. 
Correct Answer: D
Explanation/Literature review:
Erythroderma and multifocal nodules in the context of unintentional weight loss and constitutional symptoms, are most concerning for Sezary syndrome, which classically demonstrates aberrant loss of CD71. Sezary Syndrome is an aggressive subtype of cutaneous T-cell lymphoma (CTCL) that has been defined by erythroderma, superficial lymphadenopathy, and atypical T-cells in the blood1. Mycosis fungoides (MF), the most common form of CTCL, where disease is typically skin limited, patients often have a much more indolent presentation. Sezary patients on the other hand, present much more acutely and have a poorer prognosis3. The erythroderma in these patients is intensely pruritic1. Patients can also demonstrate diffuse alopecia, keratoderma, eyelid edema and dystrophic nails1. Mycosis fungoides can be difficult to diagnosis and often requires multiple skin biopsies and can mimic atopic dermatitis (AD). Our patient had a known diagnosis of AD, so it is difficult to know if this was CTCL that simply hadn’t been diagnosed yet. The prior school of thought indicated that mycosis fungoides and Sezary syndrome were on a spectrum, where Sezary was progression of a patient’s known disease1. However, while the progenitor cells in MF and Sezary both stem from memory T-cells, in MF they are considered skin resident as opposed to central resident memory T-cells in Sezary2.
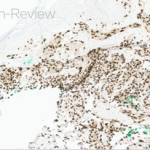
Histologic findings can range from superficial perivascular lymphocytic infiltrate, eosinophilic dermatitis with or without spongiosis, to a lichenoid infiltrate. Pautrier micro-abscesses (collections of atypical lymphocytes within the epidermis) and epidermotrophism (lymphocytes moving from the dermis into the epidermis) can also be seen3,4. Given the variability and often subtilty of histologic findings, the staining pattern is most diagnostic. The infiltrate will be CD3+ and CD4+ (answer choices B and C) but will show aberrant loss of CD7, a marker of immature T-lymphocytes (answer choice D)1. CD20 and BCL2 (answer choice A and E) would be helpful in diagnosing a B-cell lymphoma but would not be relevant in this case4. Although skin involvement is supportive of the diagnosis, to meet criteria for Sezary syndrome, patients most have an atypical, clonal T-cell proliferation within the blood. This can be identified with flow cytometry as well as T-cell rearrangement studies. The population of T-cells must have one of the following: (1) CD4+/CD8+ ratio of >10 (2) CD4+CD7- >40% or CD4+CD26- >301. Further work up includes but is not limited to CBC, CMP, peripheral blood smear and LDH, which can have prognostic importance1. Imaging is indicated to assess for lymphadenopathy as well as visceral involvement (splenomegaly, hepatomegaly)1. Importantly, Sezary patients are inherently immunosuppressed given the abnormal T-cell population as well as barrier dysfunction as it relates to erythroderma2. Sezary syndrome is deemed stage IV from skin involvement but based on lymph node and visceral involvement, more exact staging is done to best guide treatment options, with the help of our oncology colleagues5. Skin directed therapy includes topical or systemic steroids as well as phototherapy or electron beam radiation. Given blood involvement in Sezary syndrome, treatment is typically systemic1. For patients without visceral involvement, therapeutic options include extracorporeal photopheresis, oral retinoids (Bexarotene, Acitretin), interferons or histone deacetylase inhibitors (Vorinostat, Romidespin)1. Some of the targeted therapies include Brentuximab (monoclonal anti-CD30 antibody), mogamulizumab (CCR4 chemokine antibody) as well as Alemtuzumab (monoclonal anti-CD 52 antibody)1. Stem cell transplant is considered the only potentially curative option1.
Incorrect stains in answer choices3
CD20 (choice A) – B-cell antigen. Positive in B-cell lymphomas and negative in T-cell lymphomas. Target for Rituximab.
CD3 (choice B) – Pan T-cell marker. Positive in T-cell lymphomas but negative in B-cell lymphomas
CD4 (choice C) – T-helper lymphocyte marker.
BCL2 (choice E) – An oncogene that inhibits apoptosis. Use in differential diagnosis of B-cell lymphoproliferative disorders.
References:
- Sezary syndrome – statpearls – NCBI bookshelf. (n.d.) https://www.ncbi.nlm.nih.gov/books/NBK499874/
- Miyashiro D, Souza BCE, Torrealba MP, Manfrere KCG, Sato MN, Sanches JA. The Role of Tumor Microenvironment in the Pathogenesis of Sézary Syndrome. Int J Mol Sci. 2022 Jan 15;23(2):936. doi: 10.3390/ijms23020936. PMID: 35055124; PMCID: PMC8781892.
- Elston, D. M. (2019). Dermatopathology. Elsevier.
- Sézary syndrome. Pathology Outlines – Sézary syndrome. (n.d.). https://www.pathologyoutlines.com/topic/lymphomanonBsezary.html
- Olsen EA, Whittaker S, Kim YH, Duvic M, Prince HM, Lessin SR, Wood GS, Willemze R, Demierre MF, Pimpinelli N, Bernengo MG, Ortiz-Romero PL, Bagot M, Estrach T, Guitart J, Knobler R, Sanches JA, Iwatsuki K, Sugaya M, Dummer R, Pittelkow M, Hoppe R, Parker S, Geskin L, Pinter-Brown L, Girardi M, Burg G, Ranki A, Vermeer M, Horwitz S, Heald P, Rosen S, Cerroni L, Dreno B, Vonderheid EC; International Society for Cutaneous Lymphomas; United States Cutaneous Lymphoma Consortium; Cutaneous Lymphoma Task Force of the European Organisation for Research and Treatment of Cancer. Clinical end points and response criteria in mycosis fungoides and Sézary syndrome: a consensus statement of the International Society for Cutaneous Lymphomas, the United States Cutaneous Lymphoma Consortium, and the Cutaneous Lymphoma Task Force of the European Organisation for Research and Treatment of Cancer. J Clin Oncol. 2011 Jun 20;29(18):2598-607. doi: 10.1200/JCO.2010.32.0630. Epub 2011 May 16. PMID: 21576639; PMCID: PMC3422534.