
April 2023 Case Study
by Sapana Desai, MD & Azam Qureshi, MD
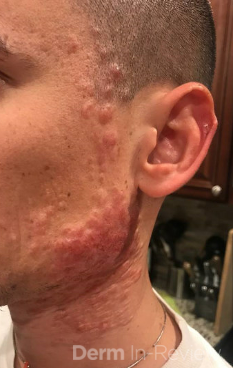
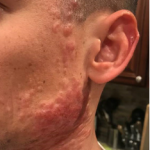
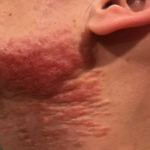
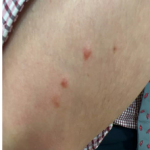
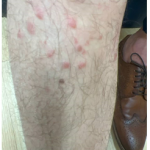
A 37-year-old male presents with a two-year history of multiple “bumps” on the face. Asymptomatic, small, pea-sized, skin-colored papules were first noted over his left lateral face and mandibular jawline. Gradually, the papules evolved into plaques, with some appearing as large as 3 centimeters. During this time, several new erythematous papules arose along the left parotid region, right elbow, and right shin, and were noted to be painful to palpation and when exposed to cold temperature. Physical examination findings are shown in Figure 1a-d.
Which of the following is commonly associated with the patient’s diagnosis?
A) Iron Deficiency Anemia
B) Renal Cell Carcinoma
C) Angiomyolipoma
D) Pheochromocytoma
E) Eccrine Spiradenoma
-
- Figure 1-a
-
- Figure 1-b
-
- Figure 1-c
-
- Figure 1-d
Correct Answer: B) Renal Cell Carcinoma
Explanation of Correct Answer:
This patient’s clinical presentation, which includes multiple erythematous, tender, dermal nodules of varying sizes in both a clustered and scattered distribution along the face and extensor surfaces of the extremities is highly suspicious for piloleiomyomas.1,2
A piloleiomyoma, regarded as the most common variant of cutaneous leiomyomas [CL] followed by angioleiomyoma, and dartoic [genital] leiomyoma, is a benign dermal neoplasm arising from smooth muscles of the erector pilorum muscle and can be solitary or multiple.1,3 Piloleiomyomas emerge with peak incidence between 20 and 40 years of age, and equally affect both genders. 90 percent of afflicted patients experience spontaneous and/or paroxysmal sharp pain often precipitated by light touch, pressure, or cold temperatures, that is believed to be attributed to cutaneous nerve compression, injury, or by contraction of tumor muscle fibers.1,2,3 While pathophysiology of piloleiomyoma remains elusive, underlying autosomal dominant [AD] inheritance of fumerate hydratase [FH] mutations are potentially associated with the development of multiple cutaneous piloleiomyoma in Hereditary Leiomyomatosis and Renal Cell Carcinoma [HLRCC] (B), also known as Reed Syndrome.4
HLRCC is a rare AD familial cancer syndrome caused by a germline amorphic allele of the FH gene, in which susceptible individuals are at increased risk for the development of cutaneous leiomyomas, early onset of multiple uterine leiomyomas, and type 2 papillary renal cell cancer.4,5 Recent meta-analyses suggest approximately 20% to 35% of individuals with germline FH mutations develop RCC and die within 5 years of diagnosis.6 Given the aggressive and metastatic nature of these tumors, annual surveillance in patients with known HLRCC should begin at 10 years of age, and surgical removal of even small sized renal tumors is recommended.4,5,
Explanation of Incorrect Answers:
Iron Deficiency Anemia (A) may be associated with Blue Rubber Bleb Nevus Syndrome [BRBNS/ Bean’s Syndrome]. Patients have characteristic cutaneous findings of multiple blue to violaceous soft and compressible nodules on the skin or mucous membranes that present at birth or in early childhood and develop numerous venous malformations involving the skin and gastrointestinal tract over their’ lifetime.7,8 Typical lesions range in size from a few millimeters to up to 4 centimeters in diameter, are rubbery in consistency, and gradually evolve with time eliciting pain most prevalent at night.8 Uniquely, lesions swell in gravity-dependent positions, and patients exhibit focal areas of hyperhidrosis overlying these lesions. Although most cases of BRBNS are sporadic, autosomal dominant inheritance has been suggested, as well as studies reporting stem cell factor/c-kit signaling systems contributing to vascular overgrowth. Lower gastrointestinal bleeding is the most common symptom ranging from obscure to massive bleeding leading to severe iron deficiency anemia. Other rare complications may include perforation, intussusception, intestinal torsion, disseminated intravascular coagulation, and thrombocytopenia.7,8
Renal Angiomyolipoma (C) is strongly associated with tuberous sclerosis complex [TSC], an AD, neurocutaneous disorder characterized by an increased predisposition to hamartoma formation, caused by genetic mutations for the proteins hamartin and tuberin, TSC1 and TSC2, respectively.9 Clinical presentation of TSC is highly variable given its multisystemic involvement, with epilepsy leading the list of neurological manifestations and accounting for 94% of affected patients, followed by cortical tubors, subependymal nodules, and subependymal giant cell astrocytomas. Frequent cutaneous manifestations encompass ash-leaf shaped hypomelanotic macules, facial angiofibroma’s, and shagreen patches.9,10 Renal angiomyolipomas may emerge in 55% to 75% of TSC patients with various longitudinal studies reporting a 75% prevalence of renal angiomyolipomas by 10.5 years of age.10,11 Although benign, TCS-associated renal angiomyolipomas are bilateral and multifocal, and when larger than 4 centimeters pose greater risks for potentially catastrophic hemorrhage when compared with isolated renal angiomyolipoma.11
Pheochromocytoma (D) may be associated with neurofibromatosis type 1- an autosomal dominantly inherited neurocutaneous disorder due to mutations in NF1 gene on 17q11.2. Pheochromocytoma is a rare neuroendocrine catecholamine-producing tumor of the adrenal medulla that synthesizes and stores excessive amounts of norepinephrine and epinephrine, which when released can evoke life-threatening cardiovascular complications.12 It may be classified as sporadic or familial, with the latter accounting for 10% of all pheochromocytoma cases, and exhibits strong association with hereditary inheritance of multiple endocrine neoplasia IIA and IIB, neurofibromatosis type 1, tuberous sclerosis, von Hippel-Lindau syndrome, Sturge-Weber syndrome, and simple familial pheochromocytoma.12,13 Up to 40% of pheochromocytomas are associated with known genetic mutations, with the most common encompassing NF-1 gene, RET proto-oncogene, VHL gene, and genes encoding succinate dehydrogenase subunits.12,13
Eccrine Spiradenomas (E) are benign dermal neoplasms closely linked with Brooke-Spiegler Syndrome [BSS], a rare AD hereditary disease which arises from heterozygous mutations in the tumor suppressor gene CYLD located on chromosome 16q12. BSS is characterized by a triad of cutaneous adnexal tumors: 1) spiradenomas- painful papules on the head, neck, and trunk, 2) cylindromas- solitary or multiple tumors of the scalp, and 3) trichoepitheliomas- skin colored papules over the central face.14,15 Eccrine spiradenomas typically emerge during late childhood or early adolescence preferentially in the head and neck region. New lesions may continue to develop throughout a patient’s lifetime. Most nodules measure 0.5 -to- 3 centimeters in diameter. Usually associated with rapid enlargement, ulceration, and bleeding, malignant transformation of either of the three tumors seen with BSS occurs in 5% to 10% of affected patients.15
References
- Kudur, MH. A Generalized Multiple Cutaneous Piloleiomyomatosis in a Young Male: Rare Care Report. Indian Journal of Dermatology. 2013; 58(3): 245.
- Albuquerque, M., Rocha, C., Costa, I., Maia, F., Sa’ Goncalves, H. Piloleiomyoma with segmental distribution- Case Report. Anais Brasileiros de Dermatologia. 2015; 90(3): 178-80.
- Bernett, C;, Mammino, J. Cutaneous Leiomyomas. 2022.
- Skala, SL., Dhanasekaran, S., Mehra, R. Hereditary Leiomyomatosis and Renal Cell Carcinoma Syndrome [HLRCC]: A Contemporary Review and Practical Discussion of the Differential Diagnosis for HLRCC-Associated Renal Cell Carcinoma. Archives of Pathology & Laboratory Medicine. 2018; 142 (10): 1202-1215.
- Ooi, A. Advances in hereditary leiomyomatosis and renal cell carcinoma [HLRCC] research. Seminars in Cancer Biology. 2020; 61(2): 158-166.
- Yu, Y., Zheng, M., Zhu, W., Zhao, F., Guan, B., Shen, Q., Yang, F., He, Q., Li, X. Hereditary leiomyomatosis and renal cell carcinoma [HLRCC]: Case series and review of the literature. Urologic Oncology. 2021; 39(11).
- Moghadam, A., Bagheri, M., Eslami, P., Farokhi, E., Asl, A., Khavaran, K., Iravani, S., Saeedi, S., Mehrvar, A., Dooghaie-Moghadam, M. Blue Rubber Bleb Nevus Syndrome because of 12 Years of Iron Deficiency Anemia in a Patient by Double Balloon Enteroscopy; A Case Report and Review of Literature. Middle East Journal of Digestive Diseases. 2021; 13(2): 153-159.
- Hu, Z, Lin, X., Zhong, J., He, Qingfang., Peng, Q., Xiao, J., Chen, B., Zheng, J. Blue Rubber Bleb Nevus Syndrome with the Complication of Intussusception: A Case Report and Literature Review. Medicine (Baltimore). 2020; 99(28).
- Zamora, E., Aeddula, Narothama. Tuberous Sclerosis. 2022.
- Portocarrero, L., Quental, K., Samorano, L., de Oliveira, Z, da Matta Rivitti-Machado, M. Tuberous Sclerosis Complex: Review Based on New Diagnostic Criteria. Anais Brasileioros de Dermatologia. October 2017; 7(4): 706-708.
- Lin, C., Jin, L., Yang, Y., Ding, Y., Wu, X., Ni, L., Yang, S., Lai, Y. Tuberous Sclerosis- Associated Renal Angiomyolipoma: A Report of Two Cases and Review of the Literature. Molecular and Clinical Oncology. August 2017; 7(4): 706-708.
- Zografos, G., Vasiliadis, G., Zagouri, F., Aggeli, C., Korkolis, D., Vogiaki, S., Pagoni, M., Kaltsas, G., Piaditis, G. Pheochromocytoma Associated with Neurofibromatosis Type 1: Concepts and Current Trends. World Journal of Surgical Oncology. 2010; 8(14).
- Naranjo, J., Dodd, S. MD, Martin, Y. MD. Perioperative Management of Pheochromocytoma. Journal of Cardiothoracic and Vascular Anesthesia. 2017; 31(4): 1427-1439.
- Mohiuddin, W., Laun, J., Cruse, W. Brooke-Spiegler Syndrome. 2018.
- Miceli, A., Ferrer-Bruker, S. Spiradenoma. 2022.