
September 2023 Case Study
by Sapana Desai, MD & Adam Rosenfeld, MD
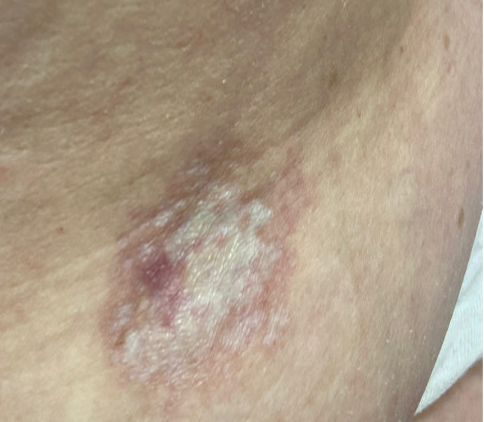
A 63-year-old woman with history of Hashimoto’s disease, diabetes mellitus, hypertension, and deep venous thrombosis presents with a progressive, multifocal, “rash” involving the extremities including the thighs and buttocks for four-to-five years. Lesions gradually advanced to affecting flexural aspects of the inguinal folds, abdomen, inframammary folds, and axillae. More recently, the patient recognized significant worsening and complains of severe pruritus, pain, and generalized discomfort. Physical examination findings are shown in Figure 1.
Which of the following diagnosis is the patient at an increased risk of developing?
A) Scleromyxedema
B) Interstitial Granulomatous Dermatitis
C) Vulvular Squamous Cell Carcinoma
D) Pheochromocytoma
E) Dermatomyositis
Correct Answer: (C) Vulvular Squamous Cell Carcinoma
Explanation of Correct Answer:
This patient’s clinical presentation, which includes multiple white atrophic plaques comprised of several flat-topped coalescing papules with maceration and scale crust, and a subsequent 4mm punch biopsy revealing a compact stratum corneum, atrophic epidermal with effaced rete, a band-like infiltrate, and associated dermal edema is indicative of extragenital Lichen Sclerosus et Atrophicus (LSetA) with vulvovaginal involvement.
LSetA is an underdiagnosed, chronic, inflammatory mucocutaneous autoimmune dermatosis with increased predilection for the anogenital sites (80% -to- 98%), but can also arise simultaneously in extragenital sites in 15% to 20% of patients- favoring the upper trunk and proximal limbs. Postmenopausal women are predominantly affected and, to a lesser extent, men, prepubertal children, and adolescents, with female-to-male ratios varying between 6:1 and 10:1.1-3 While exact etiology and pathogenesis of LSetA remains elusive, underlying genetics and family predisposition are contributory in 10% of all cases; immunological changes involving increased levels of Th1-specific cytokines, dense T cell infiltration, enhanced BIC/miR expression, as well as autoantibodies against extracellular matrix protein 1 and BP180 antigen also play integral roles. LSetA is characterized by skin atrophy and hypopigmentation, and most commonly incites clinical manifestations of severe pruritus, pain, burning, and sexual dysfunction.2,3 Its relapsing and cumbersome nature warrants every case of LSetA to be treated and managed early, as disease evolvement can lead to scarring and loss of genital anatomical architecture. Furthermore, vulvular lichen sclerosus may evolve to Vulvular Squamous Cell Carcinoma (C) in affected areas with an estimated 5% lifetime risk; however, its association with penile squamous cell carcinoma is not clear.1,3
It is imperative to note that squamous cell carcinoma develops in connection with genital, not extragenital LSetA. Literature suggests that p53 oncogenes, chronic inflammation and oxidative DNA damage are responsible for such malignant transformations. Nonetheless, the risks are significantly decreased by long-term maintenance treatment including an array of topical corticosteroids, topical calcineurin inhibitors, and well as systemic regimens for refractory cases.1,3
Explanation of Incorrect Answers:
Scleromyxedema (A) is a rare chronic cutaneous mucinosis of unknown etiology, often exhibiting association with rheumatologic diseases and monoclonal gammopathy (IgG-lambda). The disease affects adults between 30 -to- 70 years of age, with no gender predilection. Patients experience widespread symmetric eruptions of waxy, firm erythematous papules measuring 2 -to- 3cm diameter which can present close grouping in a linear distribution.1,4 Gradually, papules evolve to hardened plaques, showing marked sclerosis. In addition to cutaneous changes, 90% of patients experience plasma cell dyscrasia, in conjunction with multisystemic manifestations that may involve cardiovascular, gastrointestinal, respiratory, skeletal, muscular, renal, and nervous systems, resulting in significant morbidity and mortality. Histologically, scleromyxedema is characterized by excessive dermal mucin deposition, fibroblast proliferation, and fibrosis; still, histological findings in more than 20% of afflicted patients differs from the classic triad showing lesions that resemble interstitial granuloma annulare.4 Scleromyxedema is an unpredictable, progressive, and debilitating disease, and its relative rarity and elusive pathophysiology has precluded consensus on any one optimal treatment regimens. Even so, current literature supports aggressive treatment with high-dose intravenous immunoglobulins (IVIG) is promising while inciting the least number of side effects.
Interstitial Granulomatous Dermatitis (B) is an infrequent dermatosis that most commonly affects females, and is typically seen in the setting of rheumatic diseases [e.g. rheumatoid arthritis], but also hematological disorders, internal malignancies, infections, and antihypertensive drugs.5 Clinically, the disease is characterized by multiple erythematous papules and plaques, often with annular configuration, predominantly involving inner aspects of the limbs, lateral trunk, and intertriginous sites.5,6 The presence of cord-like indurated lesions (“rope sign”) is often considered pathognomonic. Uniquely, the histopathological examination defines the diagnosis, with presentation of dense and diffuse interstitial infiltrates within the reticular dermis- composed of histocytes in a palisade arrangement and sometimes with necrobiosis of collagen, and deposition of mucin with absence of both vasculitis and neutrophilia.1,5,6
Pheochromocytoma (D) may be associated with neurofibromatosis type 1- an autosomal dominant inherited neurocutaneous disorder due to mutations in NF1 gene on 17q11.2. Pheochromocytoma is a rare neuroendocrine catecholamine-producing tumor of the adrenal medulla that synthesizes and stores excessive amounts of norepinephrine and epinephrine, which when released can evoke life-threatening cardiovascular complications.8 It may be classified as sporadic or familial, with the latter accounting for 10% of all pheochromocytoma cases, and exhibits strong association with hereditary inheritance of multiple endocrine neoplasia IIA and IIB, neurofibromatosis type 1, tuberous sclerosis, von Hippel-Lindau syndrome, Sturge-Weber syndrome, and simple familial pheochromocytoma.8,9 Up to 40% of pheochromocytomas are associated with known genetic mutations, with the most common encompassing NF-1 gene, RET proto-oncogene, VHL gene, and genes encoding succinate dehydrogenase subunits.8,9
Dermatomyositis (E) is an inflammatory myopathy with autoimmune pathogenesis affecting women more commonly than men, with a bimodal peak of incidence between ages 5 and 14 and ages 45 and 65. Clinical presentation classically encompasses a heliotrope rash (periorbital erythema with edema, occasionally involving the cheeks and nose), Holster sign (symmetric poikiloderma of the lateral thighs below the greater trochanter), and V-sign (confluent erythematous patches over the upper central chest and lower anterior neck).1,7 Dermatomyositis is sub-classified into adult-onset disease and juvenile disease (JDM). Serum antinuclear autoantibodies are often present, as are other myositis-specific autoantibodies, which are useful as prognostic indicators while aiding in diagnosis and management of disease. Dermatomyositis is related to and possibly results from an immune-mediated process triggered by outside factors like drugs, infectious agents, and malignancy in individuals with genetic predisposition.7
References
- Bolognia, J., Schaffer, J., & Cerroni, L. (2018). Dermatology (Fourth edition.). Philadelphia,Pa: Elsevier. of clinical medicine. Retrieved August 11, 2022, from https://pubmed.ncbi.nlm.nih.gov/32516921/
- Elston, D. M. (2019). Dermatopathology. Elsevier.
- Krapf JM, Mitchell L, Holton MA, Goldstein AT. Vulvar lichen Sclerosus: current perspectives. Int J Women’s Health. (2020) 12:11–20. doi: 10.2147/IJWH.S191200
- Koronowska, S., Osmola-Mankowska, A., Jakubowicz, O., Zaba, R. Scleromyxedema: a rare disorder and its treatment difficulties. Advances in Dermatology and Allergology/Postepy Dermatol Alergol. (April 2013); 30(2): 122-126.
- Veronez, I., Luiz Dantas, F., Valente, N., Kakizaki, P., Yasuda, T., Cunha, Thais. Interstitial granulomatous dermatitis: rare cutaneous manifestation of rheumatoid arthritis. Anais Brasileiros De Dermatologia. (May 2015); 90(3): 391-393
- Coutinho, I., Pereira, N., Gouveia, M., Cardoso, J., Tellechea, O. Interstitial Granulomatous Dermatitis: A Clinicopathological Study. The American Journal of Dermatopathology. (August 2015)); 37(8): 614-619.
- DeWane ME, Waldman R, Lu J. Dermatomyositis: clinical features and pathogenesis. Journal of the American Academy of Dermatology. 2020 Feb 1;82(2):267-81.
- Zografos, G., Vasiliadis, G., Zagouri, F., Aggeli, C., Korkolis, D., Vogiaki, S., Pagoni, M., Kaltsas, G., Piaditis, G. Pheochromocytoma Associated with Neurofibromatosis Type 1: Concepts and Current Trends. World Journal of Surgical Oncology. 2010; 8(14).
- Naranjo, J., Dodd, S. MD, Martin, Y. MD. Perioperative Management of Pheochromocytoma. Journal of Cardiothoracic and Vascular Anesthesia. 2017; 31(4): 1427-1439.