June 2026 Case Study
Dillon Nussbaum, MD1
- Department of Dermatology, George Washington University School of Medicine and Health Sciences
Patient History
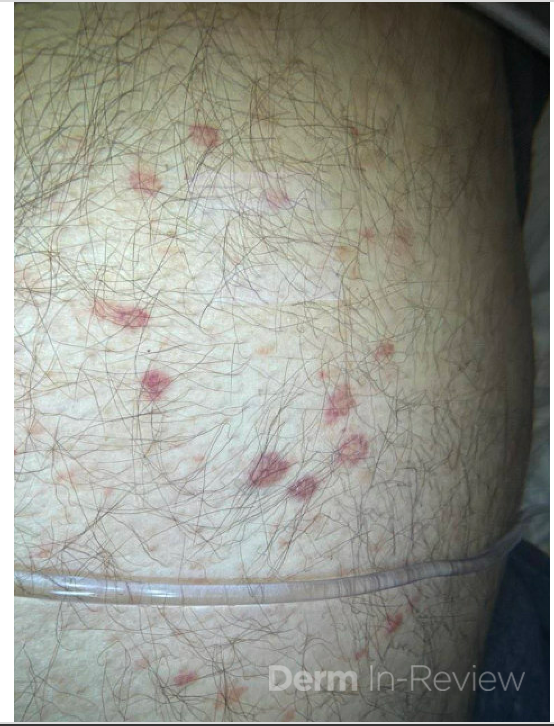
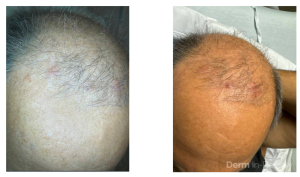
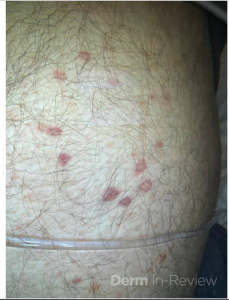
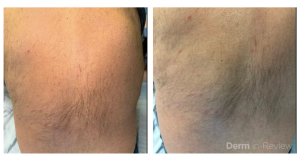
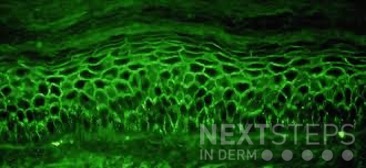
A 58-year-old woman presents with several months of painful oral erosions followed by the development of flaccid bullae on the chest and scalp. Physical examination demonstrates erosions on the buccal mucosa and a positive Nikolsky sign. Direct immunofluorescence demonstrates intercellular IgG and C3 deposition in a reticular “chicken wire” pattern throughout the epidermis (Figure 1). Due to the severity of her disease, she is started on systemic corticosteroids along with a first-line steroid-sparing agent. Which of the following best describes the mechanism by which this steroid-sparing therapy improves her disease?
Figure 1.
- Inhibition of IL-17 signaling and neutrophil recruitment
- Depletion of CD20-positive B lymphocytes, leading to reduced pathogenic autoantibody production
- Blockade of TNF-alpha mediated keratinocyte apoptosis
- Inhibition of calcineurin-dependent T-cell activation and IL-2 transcription
- Neutralization of circulating IgE antibodies bound to mast cells
Correct Answer: B
Explanation/Literature review:
This patient’s clinical presentation strongly suggests pemphigus vulgaris (PV), an autoimmune blistering disorder characterized by painful mucosal erosions, flaccid bullae, and intercellular “chicken wire” deposition of IgG and C3 on direct immunofluorescence. The pathogenesis of PV is driven by IgG autoantibodies directed primarily against desmoglein (DSG) 3 and, in many patients, DSG1. These desmosomal cadherins are critical for keratinocyte adhesion within the epidermis. Loss of adhesion between keratinocytes results in acantholysis and intraepidermal suprabasal blister formation.
Rituximab has emerged as the preferred first-line steroid-sparing therapy for moderate-to-severe pemphigus vulgaris. Rituximab is a chimeric monoclonal antibody directed against CD20, a surface antigen expressed on mature B lymphocytes. Binding of rituximab to CD20 leads to B-cell depletion through complement-mediated cytotoxicity, antibody-dependent cellular cytotoxicity, and induction of apoptosis. By reducing autoreactive B cells, rituximab decreases the production of pathogenic anti-desmoglein autoantibodies responsible for disease activity.
Multiple studies have demonstrated the efficacy of rituximab in PV. A landmark randomized controlled trial showed that rituximab combined with short-term prednisone achieved significantly higher rates of complete remission off therapy compared with prednisone alone, leading to FDA approval and adoption as first-line therapy. Subsequent systematic reviews and meta-analyses have confirmed high remission rates, reduced cumulative corticosteroid exposure, and improved long-term disease control with rituximab therapy.
The incorrect answer choices represent therapies targeting alternative immune pathways. IL-17 inhibition is used in psoriasis, calcineurin inhibition suppresses T-cell activation, TNF-alpha blockade is used in inflammatory diseases such as hidradenitis suppurativa, and anti-IgE therapy is used in allergic disease. None directly address the pathogenic B-cell–mediated autoantibody production central to pemphigus vulgaris.
References:
- Joly, P., Maho-Vaillant, M., Prost-Squarcioni, C., Hebert, V., Houivet, E., Calbo, S., Caillot, F., Golinski, M. L., Labeille, B., Picard-Dahan, C., Paul, C., Richard, M. A., Bouaziz, J. D., Duvert-Lehembre, S., Bernard, P., Caux, F., Alexandre, M., Ingen-Housz-Oro, S., Vabres, P., Delaporte, E., … French study group on autoimmune bullous skin diseases (2017). First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet (London, England), 389(10083), 2031–2040.
- Werth, V. P., Joly, P., Mimouni, D., Maverakis, E., Caux, F., Lehane, P., Gearhart, L., Kapre, A., Pordeli, P., Chen, D. M., & PEMPHIX Study Group (2021). Rituximab versus Mycophenolate Mofetil in Patients with Pemphigus Vulgaris. The New England journal of medicine, 384(24), 2295–2305.
- Murrell, D. F., & Sprecher, E. (2017). Rituximab and short-course prednisone as the new gold standard for new-onset pemphigus vulgaris and pemphigus foliaceus. The British journal of dermatology, 177(5), 1143–1144.
- Didona, D., Maglie, R., Eming, R., & Hertl, M. (2019). Pemphigus: Current and Future Therapeutic Strategies. Frontiers in immunology, 10, 1418.