Thirty Fourth Installment
Date: October 30, 2025
Guests:
Dr. Adam Friedman
Dr. Leon Tjahjono
Dr. Emily Nadelmann

Guests:
February 2026 Case Study
Caroline Clark, MD1
Patient History
A 19-year-old woman presents to dermatology clinic with a 3-year history of a progressively worsening rash on her neck, chest and hands, and nail changes. The rash is mildly itchy, has a strong odor, and worsens during the summer months when she is sweating or spending time outdoors. She has tried over-the-counter acne treatments and moisturizers without improvement.
Her father reports having similar skin problems that began in his teenage years.
Laboratory studies including complete blood count, comprehensive metabolic panel, and thyroid function tests are within normal limits.
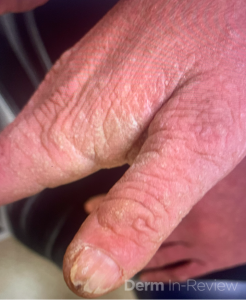

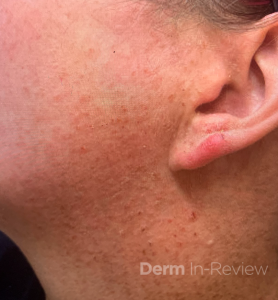
A gene mutation affecting which of the following processes is most likely responsible for this patient’s skin findings?
A) Keratinocyte proliferation and differentiation
B) Degradation of keratin through proteolytic enzymes
C) Keratinocyte Adhesion
D) Endoplasmic Reticulum calcium transport
E) Golgi Apparatus calcium transport
Correct Answer: D
Explanation/Literature Review
Darier disease is an autosomal dominant genodermatosis caused by mutations in the ATP2A2 gene, which encodes the sarco/endoplasmic reticulum (ER) calcium ATPase type 2 (SERCA2) pump (Answer choice D). This genetic defect impairs intracellular calcium homeostasis, leading to desmosome breakdown, acantholysis, and abnormal keratinization.1
The disease typically manifests in the second decade of life with greasy, yellow-brown keratotic papules and plaques in a seborrheic distribution (face, chest, back) and flexural regions, often accompanied by a characteristic malodor.1,2 Nearly all patients develop nail abnormalities including red and white longitudinal streaks with V-shaped notches at the distal edges, and many have palmoplantar pits and oral mucosal papules.
Histologically, the hallmark features are suprabasilar acantholysis with corps ronds (enlarged keratinocytes with fragmented nuclei in the spinous layer) and corps grains (small oval cells in the stratum corneum).2
The disease follows a chronic relapsing course with exacerbations triggered by heat, humidity, UV radiation, mechanical trauma, and infections.1,3
Treatment remains largely symptomatic, with oral retinoids being most effective for extensive disease, though recent evidence suggests targeting the IL-23/IL-17 axis may provide benefit in therapy-resistant cases.1,4
Explanation of Incorrect Answers
Keratinocyte adhesion (Answer choice C) is impaired in Darier disease due to ER stress and abnormal desmosome/adherens junction formation, but this is a downstream consequence rather than the primary molecular defect. Abnormal keratinocyte proliferation and differentiation (Answer choice A) are seen in psoriasis, which affects the nails with pitting, onycholysis, and subungual hyperkeratosis. The nail findings seen in onychomycosis are a result of the dermatophyte’s degradation of keratin through proteolytic enzymes (Answer choice B). Lastly, Hailey-Hailey disease (benign familial pemphigus) is caused by a mutation in ATP2C1 encoding SPCA1, a secretory pathway Ca²⁺-ATPase located in the Golgi apparatus (Answer choice D). Both Hailey-Hailey and Darier disease are calcium pump disorders with overlapping clinical features including acantholysis, however the keratotic papules and nail V-nicking are more characteristic of Darier disease.
References
January 2026 Case Study
Nathaniel Lampley, MD
Patient History
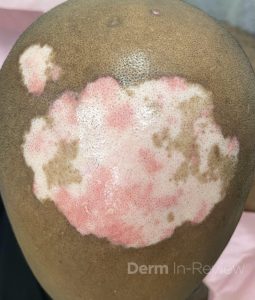
A 60-year-old male presents to your clinic for a skin cancer screening. He has no history of skin cancer or skin cancer treatments, but does admit to a heavy sun exposure history. You identify the pictured scalp lesions during your exam (Figure 1). Given the extent of scalp involvement, you prescribe a topical field therapy. A few days after beginning treatment, he develops rapidly progressive erythema, edema, erosions, and ulceration at the application sites, accompanied by fever, nausea, vomiting, and diarrhea.
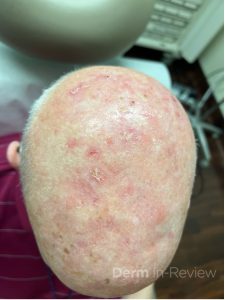
Which of the following enzymes is the patient most likely deficient in?
A) Thymidylate synthase
B) Thiopurine methyltransferase
C) Dihydropyrimidine dehydrogenase
D) Inosine monophosphate dehydrogenase
Correct Answer: C
Explanation/Literature Review
C) This patient presented with actinic keratoses (Figure 1) that were treated with topical 5-fluorouracil (5-FU). A deficiency in dihydropyrimidine dehydrogenase (DPD) can lead to impaired metabolism of 5-fluorouracil (5-FU). DPD is the rate-limiting enzyme responsible for the catabolism of over 80% of administered 5-FU. In patients with partial or complete DPD deficiency, even topical exposure can result in excessive local toxicity due to drug accumulation. 1-3
Reactions typically occur within the first several days of initiating topical therapy and can manifest as severe erosive dermatitis, ulceration, pain that is markedly more intense than the expected inflammatory response seen with standard 5-FU treatment, as well as systemic symptoms.
Recognition of this entity is important, as continued exposure can lead to severe cutaneous injury, and systemic 5-FU administration in patients with undiagnosed DPD deficiency may result in life-threatening toxicity. Management involves immediate cessation of 5-FU, supportive wound care, and avoidance of future fluoropyrimidine exposure. Genetic testing for DPD mutations may be considered.4
Explanation of Incorrect Answers
A) Thymidylate synthase is the intracellular target of 5-fluorouracil, which inhibits DNA synthesis by preventing the conversion of deoxyuridylate to thymidylate. While inhibition of this enzyme mediates the therapeutic effect of 5-FU, deficiency or alteration of thymidylate synthase does not explain excessive toxicity due to impaired drug metabolism.5
B) Thiopurine methyltransferase (TPMT) is involved in the metabolism of thiopurine medications, including azathioprine, 6-mercaptopurine, and 6-thioguanine. Deficiency in TPMT can lead to life-threatening myelosuppression with the administration of these agents, but it has no role in fluoropyrimidine metabolism.6
D) Inosine monophosphate dehydrogenase (IMPDH) is a key enzyme in de novo guanine nucleotide synthesis and is inhibited by medications such as mycophenolate mofetil. Deficiency or inhibition of this enzyme does not affect 5-FU metabolism and is unrelated to fluoropyrimidine toxicity.7
References
December 2025 Case Study
Savanna Vidal, BS1, Nagasai Adusumilli, MD, MBA1
Reviewed by: Adam Friedman, MD, FAAD1
1Department of Dermatology, The George Washington University School of Medicine and Health Sciences
Patient History
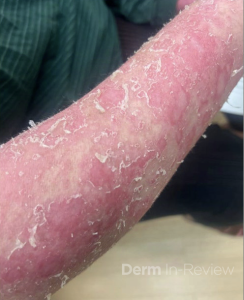
A 30-year-old female with a past medical history of major depressive disorder presented with a severe flare of a lifelong, intermittent dermatitis that had been ongoing for 3 weeks. The flare started after completing a twelve-day prednisone taper and was unresponsive to daily application of fluocinonide 0.05% cream. Throughout the lifelong management of her condition, she had previously failed management with other high-potency topical steroids, topical calcineurin inhibitors, and intramuscular corticosteroids. Family history was pertinent for similar symptoms in her identical twin sister. On examination, the patient was uncomfortable appearing and was diffusely erythrodermic with pink plaques with thick, desquamative scale distributed on the trunk and extremities (Figures 1 and 2).
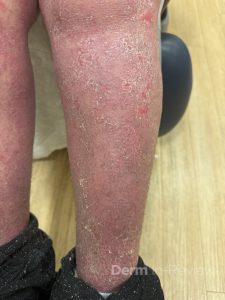
Which of the following best explains the mechanism of disease in this patient?
A. Deficient keratinocyte terminal differentiation due to filaggrin gene loss-of-function, leading to Th2-driven inflammation
B. Excessive keratinocyte-derived TSLP production, leading to eosinophil recruitment and atopic diathesis
C. Autoantibody formation against desmoglein 1 and 3, causing intraepidermal acantholysis and inflammatory cascades
D. Unregulated epidermal protease activity due to loss of LEKTI inhibition, resulting in barrier dysfunction and upregulation of Th2 and Th17 pathways
E . Gain-of-function mutation in STAT3 leading to enhanced Th17 differentiation and chronic mucocutaneous candidiasis
Correct Answer: D
Explanation/Literature Review
Netherton syndrome (NS) is a rare autosomal recessive genodermatosis caused by loss-of-function mutations in the SPINK5 gene that encodes the serine protease inhibitor LEKTI (lymphoepithelial Kazal-type-related inhibitor). As LEKTI normally functions to suppress epidermal kallikrein (KLK), particularly KLK5 and KLK7, skin barrier integrity is disrupted and a complex immunological cascade follows, characterized by heightened Th2 and Th17 inflammatory pathways.1,2
Clinically, NS is defined by the classic triad of ichthyosis linearis circumflexa, trichorrhexis invaginata, and atopic diatheses.1-3 While the overlap of NS with an atopic dermatitis-like presentation may lend itself to a Th2-predominant phenotype, recent molecular profiling has identified significant upregulation of Th17-related cytokines in the erythrodermic variant of NS.2 Pathologic evaluation is nonspecific, exhibiting hyperkeratosis, acanthosis, focal hypergranulosis, and a superficial perivascular lymphocytic infiltrate. Immunohistochemical staining for LEKTI may further help with diagnosis, though genetic testing is required for confirmation.1, 4
Topical anti-inflammatory agents, notably corticosteroids, are mainstays in the management of NS, though the increased absorption secondary to the skin barrier defect present in these patients must be considered.1,5 Biologic agents have emerged as promising treatments at the case report level, targeting specific inflammatory axes in NS. Dupilumab, an IL-4Rα antagonist that inhibits IL-4 and IL-13 signaling, has demonstrated variable clinical efficacy in multiple case series, with an ongoing clinical trial further investigating its efficacy.6 In the erythrodermic phenotype of NS with IL-17 and IL-36 predominant-pathways, IL-17 inhibitors secukinumab and ixekinumab have shown remarkable improvement in multiple patients.7-8 The role of other biologics such as TNF-α inhibitors (e.g. infliximab), ustekinumab (IL-12/23 blockade), anakinra (IL-1R antagonist), and omalizumab (anti-IgE) has been explored, but responses remain inconsistent, likely reflecting the heterogeneity of NS immunophenotypes.9 There are no FDA-approved treatments for NS, so all management options mentioned are off-label and may require additional effort from the prescribing clinician to help patients access the medication.
Incorrect Options
A. Filaggrin deficiency is the hallmark of ichthyosis vulgaris and a major predisposing factor for atopic dermatitis, but not for NS. The pathogenesis in NS is centered on protease dysregulation.10
B. TSLP is a key cytokine in AD and promotes Th2 polarization and eosinophil recruitment, but it is not the primary abnormality in NS.11
C. This describes pemphigus vulgaris, an autoimmune blistering disorder unrelated to NS.12
E. STAT3 gain-of-function mutations are associated with Job syndrome (hyper-IgE syndrome) and chronic mucocutaneous candidiasis. While both Job syndrome and NS involve elevated IgE, the molecular pathogenesis and cytokine profile differ significantly.13
References
Robin Picavia, MD1
Patient History
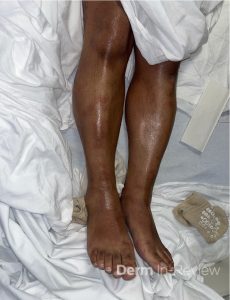
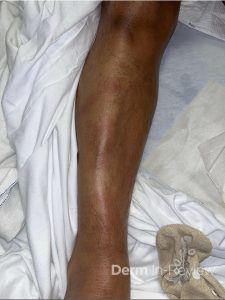
A 52-year-old man presents with 2 weeks of fever, bilateral ankle swelling, and painful erythematous nodules on his shins. He reports a month of fatigue, decreased appetite, and 10-lb weight loss. Examination shows tender red subcutaneous nodules on the anterior lower legs and swelling of both ankles. Labs show an elevated ESR. Chest CT demonstrates bilateral hilar lymphadenopathy with mediastinal adenopathy and a small right upper lobe nodule. Biopsy of a shin lesion is consistent with erythema nodosum.

Question:
Which of the following is the most likely diagnosis?
A. Löfgren syndrome
B. Polyarteritis nodosa
C. Erdheim–Chester disease
D. Erythema induratum (nodular vasculitis)
E. Sweet syndrome
Correct Answer: A. Löfgren syndrome
Explanation/Literature review:
Löfgren syndrome (Answer A) is an acute, self-limited presentation of sarcoidosis characterized by the classic triad of erythema nodosum, bilateral hilar lymphadenopathy, and acute
symmetric polyarthritis, most commonly involving the ankles. 1 Patients typically present with fever, malaise, and fatigue, often accompanied by weight loss and night sweats. 1 The arthritis is usually abrupt in onset, tender, and may be associated with periarticular inflammation or ankle swelling. Erythema nodosum appears as tender, erythematous nodules on the anterior shins and represents a reactive septal panniculitis rather than granulomatous skin disease. Chest imaging reveals bilateral hilar and right paratracheal lymphadenopathy; parenchymal lung involvement is less common in the acute setting. 1 Laboratory findings may include elevated ESR/CRP, mild leukocytosis or lymphopenia and occasionally elevated liver enzymes.
Importantly, Löfgren syndrome is associated with an excellent prognosis, with spontaneous remission in up to 80–90% of patients within 6–12 months. Treatment is often supportive, including NSAIDs for arthralgia, while short courses of corticosteroids may be used in more severe cases; long-term immunosuppression is rarely needed. Because the triad is highly specific, tissue biopsy is not always required unless atypical features or alternative diagnoses are suspected. 1
Polyarteritis nodosa (Answer B) is unlikely since it is a medium vessel necrotizing vasculitis that presents with livedo reticularis, neuropathy, renal involvement, and systemic symptoms but does not produce septal panniculitis or bilateral hilar lymphadenopathy. 2 Erdheim-Chester disease (Answer C) typically manifests with long-bone sclerosis, retroperitoneal fibrosis, orbital infiltration, and foamy histiocytic infiltrates, none of which fit this patient's biopsy-proven erythema nodosum or acute ankle arthritis; it also does not present with the sarcoidosis triad. 3
Erythema induratum (Answer D) involves lobular panniculitis with vasculitis, often associated with tuberculosis, and usually presents with tender nodules on the posterior calves rather than anterior shin lesions, making it inconsistent with this patient's clinical and histologic findings. 4
Sweet syndrome (Answer E) presents with acute tender plaques and a dense neutrophilic infiltrate on biopsy rather than septal panniculitis and does not produce bilateral hilar
lymphadenopathy, further excluding it from consideration. 5 Taken together, the clinical features and biopsy results most strongly support acute sarcoidosis consistent with Lofgren
syndrome.
References
1. Haimovic, A., Sanchez, M., Judson, M. A., & Prystowsky, S. (2012). Sarcoidosis: A
comprehensive review and update for the dermatologist. Journal of the American
Academy of Dermatology, 66(5), 699.e1–699.e18.
2. Stewart, M., & Mohan, N. (2022). Cutaneous polyarteritis nodosa diagnosis and
treatment: A retrospective case series. Journal of the American Academy of
Dermatology, 87(6), 1370–1373.
3. Maredia, H., et al. (2024). Atypical cutaneous manifestation of Erdheim–Chester disease
successfully treated using medical and laser therapy. JAAD Case Reports, 64, 75–78.
4. Gilchrist, H. and Patterson, J.W. (2010), Erythema nodosum and erythema induratum
(nodular vasculitis): diagnosis and management. Dermatologic Therapy, 23: 320-327.
https://doi.org/10.1111/j.1529-8019.2010.01332.x
5. Heath, M. S., & Ortega-Loayza, A. G. (2015). Erythema nodosum and other
panniculitides: Classification and approach. Journal of the American Academy of
Dermatology, 73(5), 691-705.
Caroline Clark, MD1
Patient History
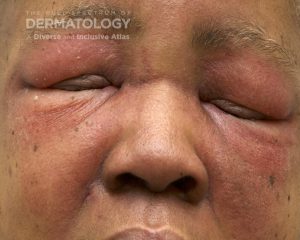
A 69-year-old male with a history of hypertension, rheumatoid arthritis, and HIV (recent viral load undetectable), presents in dermatology clinic with purplish discoloration around both eyes/upper cheeks which he states developed around 1 month ago (figure 1). He endorses vague upper extremity muscle pain, but otherwise review of systems is negative. A punch biopsy was performed which vacuolar interface dermatitis with dermal mucin deposition.
Question:
Next, appropriate bloodwork is performed and reveals this patient has an elevated creatine kinase and positive Anti-TIF1-γ antibodies. What is the next best step in management?
Answer: 2. Age-appropriate cancer screening + whole body PET scan
Explanation/Literature review:
Dermatomyositis (DM) is an idiopathic inflammatory myopathy characterized by progressive, symmetric, proximal muscle weakness and an array of distinct cutaneous manifestations. Classic skin findings include a heliotrope rash, periorbital edema, red or violet papules overlying the bony prominences on the dorsal hands and other joints, such as the elbows and knees (Gottron papules), red-purple rash on the upper chest (V-sign), upper back, shoulders, and neck (shawl sign), or on the lateral hips and upper thighs (holster sign), and characteristic nail findings.1,2
Initial workup for DM includes a detailed history and full body skin examination, baseline laboratory testing with muscle enzymes (creatine kinase, aldolase) and autoantibody testing. A muscle biopsy may be indicated but should be reserved for equivocal cases in which diagnosis is unclear (Answer 3).
DM is associated with an increased prevalence of malignancy. Cancer risk is highest within a year of diagnosis and remains elevated for around 5 years.1 Certain myositis-specific autoantibodies (MSAs) are present in about 20% of patients with DM.2 MSAs are associated with varying degrees of malignancy risk and, if positive, can help guide appropriate management of cancer screenings. Anti-transcription intermediary factor 1 gamma (anti-TIF1- γ) and anti-nuclear matrix protein 2 (anti-NXP2) antibodies are both associated with high risk for DM-associated malignancy.1 Individuals who are positive for either of these antibodies should undergo both age-appropriate cancer screening in addition to whole body imaging with either CT, MRI or PET (Answer 2).1 Adult patients with DM who are negative for both anti-TIF1- γ and anti-NXP2 have a lower risk of malignancy associated DM and do not require additional cancer workup outside of standard age-based cancer screening, as more invasive cancer screening is unlikely to improve outcomes (Answer 1).
In all DM patients, regardless of antibody status, there should be a low threshold to undergo standard screening with pulmonary function testing (PFTs) and DLCO. Anti-melanoma differentiation-associated protein 5 (MDA5) antibody-positive DM is associated with increased risk for rapidly progressive interstitial lung disease (ILD). Patients positive for this antibody, or those with concerning respiratory symptoms, warrant additional workup with high-resolution CT scan (Answer 4) and pulmonology referral.3
While IVIG (Answer 5) is among the treatments available for DM, this is traditionally not initiated as a first-line therapy and cancer screening is the first priority in this patient.2
References:
Savanna Vidal, BS1, Nagasai Adusumilli, MD, MBA1
1Department of Dermatology, The George Washington University School of Medicine and Health Sciences
Patient History
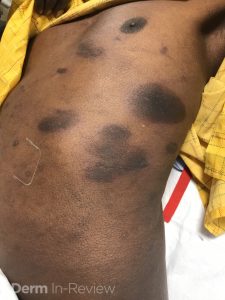
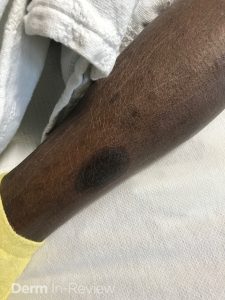
A gentleman in his 60s with PMH of HIV (CD4 178 on antiretroviral therapy), Hep C, Hep B, HTN, and prediabetes, was admitted for hypoglycemia and hypothermia after being found down. Dermatology was consulted for a diffuse pruritic, burning rash on the trunk and extremities for about 6 months. Medium potency topical corticosteroids provided partial relief of symptoms, but the patient described increasing number of pruritic plaques. Medication review showed amlodipine, lisinopril, trimethoprim/sulfamethoxazole, and metformin for his chronic medical conditions. Figures 1 and 2 demonstrate the pertinent skin findings.
Which of the following best describes the expected histopathology findings?
Correct Answer: D
Explanation/Literature Review
With a CD4 count less than 200, this patient was newly taking trimethoprim/sulfamethoxazole for prophylaxis against pneumocystis pneumonia (PCP). The multiple, sharply demarcated, mostly circular and ovoid, thin, pigmented, pruritic plaques are most consistent with fixed drug eruption likely secondary to trimethoprim/sulfamethoxazole. The characteristic histopathology findings for fixed drug eruption include the mismatch of an acute stratum corneum and the chronic changes of the superficial dermis. The interface dermatitis is more vacuolar rather than lichenoid with a diffuse band of inflammation obscuring the dermal-epidermal junction. Eosinophils and pigment incontinence are commonly seen (Choice D).1 The histopathologic findings can be similar to erythema multiforme and graft-versus-host disease. However, both histopathologic differential diagnoses will typically have more necrotic keratinocytes.1
While theoretically any medication can cause a fixed drug eruption, trimethoprim/sulfamethoxazole is a classic offender estimated to encompass the majority of cases.2 In addition to the clinical morphology, dermatologists must be aware of common contexts for which classic offending medications may be prescribed. Tetracyclines are often implicated for fixed drug eruption on the penis, and pseudoephedrine is a common cause of the nonpigmented variant.2 After the initial exposure, symptoms between a few days to a week is the typical time course. However, the symptoms can recur sooner and with more severity with repeated subsequent exposures.2 After the appropriate diagnosis of fixed drug eruption, this patient’s PCP prophylaxis was switched to atovaquone, trimethoprim/sulfamethoxazole was added to his drug allergy list, and triamcinolone 0.1% ointment alleviated symptoms of pruritus.
Incorrect Options
References
Author: Alexis E. Carrington, MD
Reviewed by: Anna Yasmine Kirkorian, MD
A 5-month-old male with no significant medical history presents with a 3-month history of a generalized pruritic rash. The eruption began on the feet and spread upward. The patient’s mother reports significant sleep disruption due to itching. He failed oral valacyclovir and topical desonide previously prescribed. The patient’s mother and sister also developed pruritic rashes.
On examination, the patient had generalized inflammatory nodules and pustules, most prominent on the axillae, trunk, nape of the neck, and lateral feet.
Which of the following is the most appropriate initial diagnostic test for this patient?
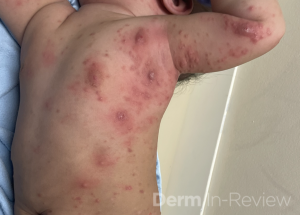
Image 1: Many scattered erythematous-violaceous nodules extending from the left inner arm to the trunk.
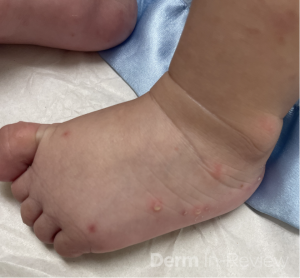
Image 2: Several scattered pustules and inflammatory nodules on the lateral and dorsal left foot.

Image 3: Positive mineral oil prep demonstrating a live scabies mite and scybala.
A) Skin mineral oil preparation
B) Bacterial culture
C) Skin biopsy for histopathology
D) Fungal culture
Correct Answer: A) Skin mineral oil preparation
A mineral oil prep is the first-line, rapid, and inexpensive test to diagnose scabies by directly visualizing mites, eggs, or scybala under the microscope. Skin biopsy and cultures may be considered if the diagnosis remains uncertain after initial evaluation but are not the first step when scabies is strongly suspected based on clinical findings.
This patient has scabies confirmed by positive mineral oil prep. Scabies is caused by infestation with the mite Sarcoptes scabiei and presents with intensely itchy papules, nodules, and burrows, often worsening at night. Scabies is highly contagious via skin-to-skin contact and fomites.1,2 Scabetic nodules on the trunk, axillae and genitalia are the most common presentation in infants and can remain for several months after resolution.3
A skin mineral oil preparation (Choice A) is the most appropriate and efficient initial diagnostic test for confirming scabies by directly visualizing mites, eggs, or scybala under the microscope. A bacterial culture (Choice B) would only detect secondary bacterial infections, such as impetigo from scratching, and would not diagnose the underlying scabies infestation. A skin biopsy (Choice C) could potentially reveal mites or burrows under histopathology, but it is more invasive, time-consuming, costly, and less sensitive early in the disease. A fungal culture (Choice D) would be reasonable if considering deep fungal infections like coccidioidomycosis or cryptococcosis, but would be less likely to be seen in an immunocompetent patient.
All household members should be treated simultaneously to prevent reinfestation, and thorough cleaning of bedding, clothing, and fomites is crucial.
References:
Author: Dillon Nussbaum, MD1
Patient History
A 42-year-old man presents with several well-demarcated, erythematous plaques with central atrophic hypopigmentation, peripheral hyperpigmentation, and adherent scale on his scalp, back, and cheeks for several months (Figure 1). He notes mild pruritus and hair loss in the affected areas but denies systemic symptoms. A punch biopsy is obtained and is significant for interface dermatitis and mucin deposition.
Which of the following ophthalmologic findings may develop in a small percentage of patients taking the first-line systemic therapy for this condition longitudinally?
Correct Answer: C
Explanation/Literature Review:
Histopathologic findings of interface dermatitis and mucin deposition along with clinical findings of atrophic hypopigmented plaques with peripheral hyperpigmentation are consistent with a diagnosis of discoid lupus erythematosus (DLE).1 The first-line systemic agent for widespread or refractory DLE is hydroxychloroquine, which inhibits Toll-like receptors 7 and 9 and downregulates the interferon mediated inflammatory cascade implicated in DLE. Although generally well-tolerated, hydroxychloroquine carries a risk of retinal toxicity, particularly with long-term use or dosing above 5 mg/kg/day. Hydroxychloroquine accumulates in the retinal pigment epithelium and can cause a central scotoma, which presents as a focal blurry or blind spot at the center of the visual field, and is typically irreversible. Studies indicate the incidence of central scotoma in patients taking hydroxychloroquine ranges from 0.5% to 7.5% after 5 years of therapy, with most cases being mild, and the variability depending on diagnostic criteria. Baseline and annual ophthalmologic exams, including visual field testing and optical coherence tomography, are recommended after 5 years of therapy or earlier in higher-risk patients.2-4
DLE commonly occurs in sun exposed areas with a predilection for the conchal bowls, often resolves with scarring and alopecia, and is worsened by tobacco use. Topical therapies include corticosteroids and non-steroidal anti-inflammatory agents in addition to daily photoprotection. Hydroxychloroquine is first line for both systemic lupus erythematosus (SLE) and DLE and other systemic immunosuppressants are frequently used in conjunction with hydroxychloroquine when appropriate. Janus kinase inhibitors are increasingly being utilized off label for both DLE and SLE. While uncommon, DLE can be associated with SLE, and if so, monoclonal antibodies such as belimumab or anifrolumab can be utilized.5,6 Incorrect answers include Kayser-Fleischer rings, which are dark copper colored rings on the periphery of the iris seen in Wilson’s disease. Cataracts, or clouding of the lens, is associated with corticosteroid use among other etiologies, but has not been associated with the use of hydroxychloroquine. Retinal detachment and optic neuritis are unrelated to hydroxychloroquine use.
References:
Author: Nikita Menta, BA1, Nidhi Shah, MD1, Karl Saardi, MD1
Patient History
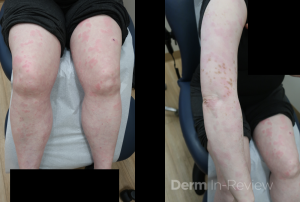
A 43-year-old female presented to the clinic with several painful, erythematous, edematous plaques in addition to hyperpigmented plaques on the arms and legs, which had been present for more than 24 hours. The rash initially started about four years prior to presentation and was unresponsive to a variety of oral antihistamines and a four-month trial of omalizumab. The patient also endorsed intermittent episodes of angioedema as well as occasional low back and knee pain. She previously underwent a punch biopsy on the lower left back; hematoxylin and eosin staining demonstrated marked dermal edema, margination of neutrophils with perivascular and interstitial infiltrate including eosinophils, perivascular nuclear debris with neutrophils, and significant extravasation of erythrocytes. Exam findings are shown in Figure 1.
Based on the clinical presentation and biopsy findings, which of the following laboratory tests would be most useful in determining the prognosis of this condition?
A) Antinuclear antibodies
B) Erythrocyte sedimentation rate
C) Complement Levels
D) C-reactive protein
E) Liver function tests
F) Rheumatoid factor
G) Antithyroid antibodies
Correct Answer: C
Explanation/Literature Review:
This patient’s treatment-resistant urticarial plaques combined with histopathologic findings demonstrating perivascular nuclear debris with neutrophils and extravasation of erythrocytes are consistent with a diagnosis of urticarial vasculitis (UV). UV classically presents as recurrent, long-lasting wheal-like lesions with a predilection for the trunk and proximal extremities, accompanied by histopathologic findings of leukocytoclastic vasculitis.1 UV can present with or without angioedema. Additionally, features that help distinguish UV from chronic spontaneous urticaria (CSU) include the persistence of lesions beyond 24 hours, predominant symptoms of burning and pain compared to pruritus, and residual post-inflammatory purpura or hyperpigmentation upon healing.2,3 UV is a rare condition with an estimated incidence of 0.5 per 100,000 in the United States.4 It most commonly occurs in individuals in their 5th to 7th decades of life and demonstrates a female gender predominance.5 Regarding pathophysiology, UV is a type III hypersensitivity reaction; however, in most cases, the triggering antigen is often unidentified.6 While the majority of UV cases are idiopathic, a small percentage of cases have been associated with medications, infectious diseases, malignancies, and autoimmune diseases, commonly systemic lupus erythematosus.2
There are two main subtypes of UV, which are classified based on complement levels, the most important indicator of UV prognosis (answer choice C). Patients with normal complement levels, or normocomplementemic UV (NUV), tend to have skin-limited vasculitis. Conversely, patients with low complement levels, or hypocomplementemic UV (HUV), are much more likely to experience systemic disease.1,2 The most common systemic manifestations of HUV include arthralgia, arthritis, inflammatory eye diseases, pulmonary symptoms (mimicking asthma or COPD), gastrointestinal symptoms, and renal pathologies (proteinuria/hematuria).1 Finally, there is also a more severe subtype of HUV, called HUV syndrome (HUVS), which is defined by multiorgan involvement.1
UV is a notoriously challenging condition to treat, further complicated by the lack of standardized guidelines and randomized controlled trials. In a recent review article, H1-antihistamines, omalizumab, and immunomodulators were among the treatments with the most robust evidence in UV management.4,7 However, while H1-antihistamines were widely employed, they were only effective in improving skin lesions in 9.9% of 141 patients.7 Omalizumab demonstrated much greater efficacy, with 76% of 108 patients experiencing improvement in cutaneous symptoms and many also experiencing improvement in systemic symptoms such as arthralgia and abdominal pain.7 Among immunomodulators, systemic corticosteroids, dapsone, and hydroxychloroquine had the most substantial and strongest evidence supporting their use in addressing cutaneous and systemic symptoms of UV; however, the potential adverse effects of these drugs must be carefully considered.4,7 The aforementioned review article proposed a treatment algorithm recommending that mild UV should be approached like CSU, starting with H1-antihistamines and escalating to omalizumab, whereas moderate-to-severe UV should be managed with corticosteroids and/or other immunomodulatory agents depending on the patient profile.7 While this algorithm offers some direction, there is a critical need for high-quality, head-to-head trials to determine the optimal approach to UV management.
Incorrect answer choices:
The other laboratory tests are not indicative of prognosis but are warranted in the workup of patients with UV, particularly HUV. Inflammatory markers, specifically erythrocyte sedimentation rate (B) and C-reactive protein (D), are typically elevated in UV.1,5 Additionally, since UV can be associated with autoimmune diseases, autoimmune laboratory tests, including but not limited to antinuclear antibodies (A), rheumatoid factor (F), and anti-thyroid antibodies (G) are often included in the workup to detect or rule out underlying autoimmune diseases.5 Finally, while liver dysfunction is not a common systemic manifestation of UV, liver function tests (E) may be included alongside standard laboratory tests, such as complete blood count and basic metabolic panel, in the workup of UV.5
References
© 2002-2026 Derm In-Review