Case Studies are a resource of Derm In-Review,
brought to you by our educational partner
Featured Case Study

{kind=link}
{kind=link}
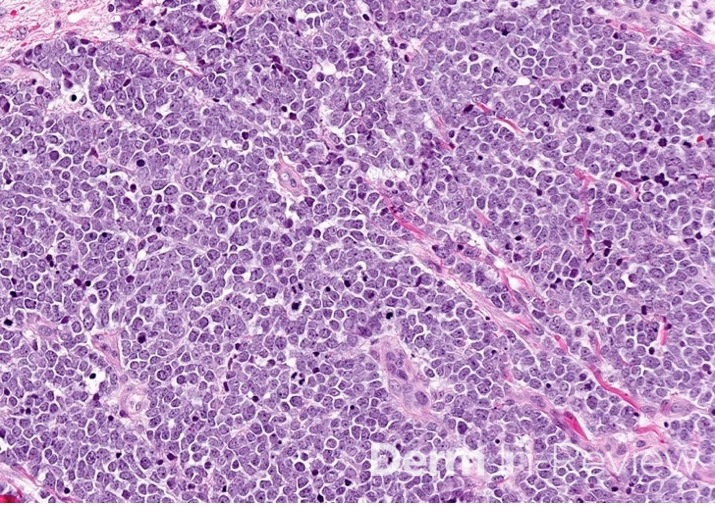
Credit: Dillon Nussbaum, MD1
Department of Dermatology, George Washington University School of Medicine and Health Sciences

Case Studies are a resource of Derm In-Review,
brought to you by our educational partner
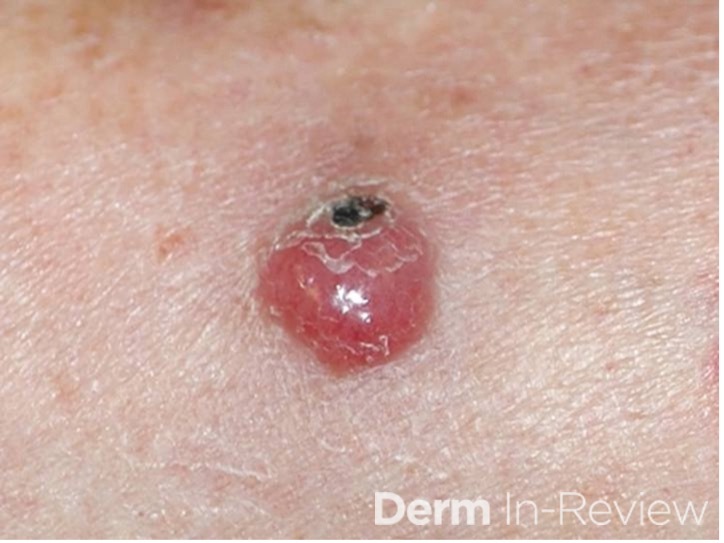
Credit: Dillon Nussbaum, MD1
Department of Dermatology, George Washington University School of Medicine and Health Sciences
© 2002-2026 Derm In-Review